Some thoughts on omega-6 depletion, and the success of low-fat diets for it
-
I tend to write a lot, so I've separated the subject into topics so you can only go to what interests you, a sort of “tl;dr” is in the titles themselves and I have no interest in summarizing any further. The aim was to be brief and I think it is, considering the complexity of the subject, just not for reddit haha
On EFA-deficiency
- Fatty acid composition in essential fatty acid deficiency(EFAD): What happens to fatty acids when you deplete omegas-6?
- Unsaturation index(UI): In the absence of external unsaturation (MUFAs and PUFAs from diet), the body increases its own capacity to desaturate SFAs and MUFAs
- D9 desaturase activity: Why is avoiding MUFA while depleting PUFA almost always not worth the trouble?
On Low-fat
- Low fat(HCLF): Basically a high MUFA diet
- Low fat increases DNL, but why does it increase even more as PUFA levels drop?(DNL gets a new role! Our poor overworked employee)
Fatty acid composition in EFAD: What happens to fatty acids when you deplete omegas-6?
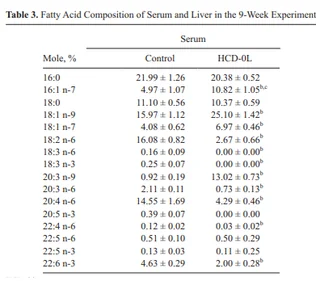
The consequence of a diet lacking in essential fatty acids is predominantly a depletion of omega-6 PUFAs (although omega-3 PUFAs to a lesser extent), and a considerable increase in MUFAs (16:1n-7, 18:1n-9) and derivatives (20:3n-9). This profile, in general, occurs in diets deficient in essential fatty acids, even those that add a fat source such as hydrogenated coconut oil.
Unsaturation index(UI): In the absence of external unsaturation (MUFAs and PUFAs from diet), the body increases its own capacity to desaturate SFAs and MUFAs.
I think that intuitively everyone knows that for a cell to be viable it can't be 100% saturated or unsaturated, and the consequence of not bringing this information to the “conscious” is to not consider that this means that the body maintains control over the ratio of unsaturated to saturated of a cell (and an entire tissue, for that matter).
And what does this mean? It means that the body's natural response to PUFA depletion (i.e. the response to drastically reduced unsaturation) is to increase MUFAs, and when the absence of PUFAs is severe, MUFAs begin to be desaturated to PUFAs, as is the case with Oleic to Mead Acid. This is the only way the body can meet at least the minimum level of unsaturation that a cell/tissue requires without external aid.
Feeding diets rich in saturated fatty acids decreases the ratio of n6/n3 PUFA in PL of mitochondria from rat liver, heart, or kidney, but leaves the ratio of saturated/unsaturated fatty acyls unchanged, suggesting a 'homeostatic' mechanism to Gibson et al. Such homeostasis, like the increased omega 9-unsaturation, may provide membrane fluidity for some nonspecific systems and prevent lethal outcome of EFA-deficiency.
Frederick L. Hoch. LIPIDS AND THYROID HORMONESIncreased Unsaturation Index = Increased pufa(fluidity) to SFA(rigidity?)
This also means that if there is a min and a max of unsaturation, providing more of one inevitably reduces more of the other. More PUFAs means less MUFAs; more omega-6 means less MUFAs/omega-3; more omega-3 means less MUFAs/omega-6; more MUFAs means less PUFAs.
However, they blocked plasma membrane lipid reactive oxygen species (ROS) accumulation and reduced PUFA incorporation into phospholipids.
Their work suggests that exogenous MUFAs may alter cell membrane properties by displacing PUFAs such as AA, EPA, and DHA. Das, U. N. (2019).
Saturated Fatty Acids, MUFAs and PUFAs Regulate Ferroptosis.D9 desaturase activity: Why is avoiding MUFA while depleting PUFA almost always not worth the trouble?
The consequence of depleting PUFAs from the diet is the maximization of endogenous desaturation systems, max activity of D9D, D6D, D5D and elongases. There is no other response than that to maintain at least the minimum level of unsaturation viable for a cell/tissue when there is not a sufficient external source of unsaturation.D6D and D5D are fully induced only in EFA-deficient conditions, and are suppressed when adequate precursor PUFAs are supplied from the diet (10, 11).>
SCD-1 is expressed constitutively in adipose tissue and is markedly induced in liver in response to feeding with a high-carbohydrate diet (122)
Nakamura, M. T., & Nara, T. Y. (2004). STRUCTURE, FUNCTION, AND DIETARY REGULATION OF Δ6, Δ5, AND Δ9 DESATURASES.I'll comment later, but I don't think the trigger for SCD1(pufa depletion context) in the liver is carbohydrates per se, but rather how the liver senses the increase in DNL SFAs in relation to the Unsaturation Index.
On low-fat
Low fat: Basically a high MUFA diet
Carbs are used immediately, stored in the form of glycogen and if glycogen has reached its full capacity (or the body has reached the physical limit of processing) DNL is triggered to deal with the excess. I think the SCD1 trigger in these cases is not the carbs or SFAs per se, but rather the oversaturation provided by DNL pushing the Unsaturation Index down and activating a feedback system to return to homeostasis, increasing unsaturation with MUFAs.It seems to be the case:
Brenner, R. R., Castuma, C. E., & Garda, H. (1986). Possible mechanisms by which microsomal lipid bilayer composition modify bound enzyme kinetics
The effect of fluidity on the desaturating enzymes is evoked in the opposite direction: the desaturation decreases with the increase of fluidity. If unsaturated acids increase fluidity, a decrease of unsaturated acid biosynthesis by increased fluidity would implicate the existence of a self regulatory mechanism in the membrane.
Garg, M. L., Wierzbicki, A. A., Thomson, A. B. R., & Clandinin, M. T. (1988). Dietary cholesterol and/or n − 3 fatty acid modulate Δ9-desaturase activity in rat liver microsomes
The reduction in D9-desaturase activity was significantly higher for animals fed diets containing fish oil (90% lower than observed for animals fed the beef tallow diet) compared with the linseed oil diet (60% reduction).
Therefore, feeding of the linseed oil and fish oil diets may have increased membrane fluidity and correspondingly decreased D9-desaturase activity in a tendency to maintain physicochemical properties
These results are in agreement with previous studies which found that D9-desaturase is regulated by the overall unsaturation of membrane phospholipids which can be altered by changes in dietary fat.
New cells or cells that need to renew their unsaturated fatty acids will replace theirs with the available unsaturated fatty acids, the constant DNL>SCD1 trigger makes plenty of MUFAs available and this is what they will use to build/renew if MUFA fulfills the function. The MUFAs “dilute the LA” in the OQs probably for this reason, but with patience they also accelerate the depletion of omegas-3/6 from all other tissues as a matter of competition, adipose tissue that acts as a reservoir will be the most resistant and will continue to influence the results until it is diminished or the rate of renewal finally replaces the stored PUFAs.
The relationship between the Unsaturation Index and DNL>SCD1 seems to be much more responsible for the success of low-fats in depleting LA compared to ketogenic than the total amount of LA. I believe that even if a HCLF ingests the same amount of LA (or even more) compared to a ketogenic equivalent, it would still objectively have much less LA, with a lot of MUFA you can simply get away with a little more LA (now add that to HCLF's ability to also keep total LA very low).
Having said all that, I would expect the places with the greatest lipogenic capacity to be the first to show a deficiency of EFAs for precisely these reasons(DNL pushing local Unsaturation Index down), and apparently that's what happens, at least in animals. You can even see that the need to normalize the unsaturation index is so great that stearic is almost depleted as well, despite the crazy level of DNL that is taking place. Regarding the article “FATTY ACID SYNTHESIS DURING FAT-FREE REFEEDING OF STARVED RATS”, the liver is the first place to be considered EFA-deficient.
Normal -> Fasting+refeed(fat-free)
Liver
Stearic(18:0): 24.9 -> 6.0
Palmitoleic(16:1): 1.4 -> 12.1
Oleic(18:1): 13.3 -> 36.4
Linoleic(18:2): 16.6 -> 0.1
ARA(20:4): 4.9 -> 0.1Heart(it's not the same as the liver, the change is more about how the heart interacts with FFAs)
Palmitoleic(16:1): 1.0 -> 2.0
Oleic(18:1): 9.65 -> 18.0
Linoleic(18:2): 29.8 -> 18.0
ARA(20:4): 10.4 -> 11.9Adipose
Palmitoleic(16:1): 5.1 -> 6.8
Oleic(18:1): 37.6 -> 42.5
Linoleic(18:2): 23.3 -> 19.0Low Fat increases DNL, but why does it increase even more as PUFA levels drop?(DNL gets a new role! Our poor overworked employee)
It is also possible that with the progression of EFA deficiency, DNL takes on a new role which is to provide substrate for unsaturation (SCD1>MUFAs>omega-9 PUFAs) and will remain high as long as unsaturation is not adequately normalized. In this case SCD1 would become the DNL trigger and not the other way around.It's hard to know with 100% certainty, but I'm confident
Jeffcoat and James [81] who demonstrated that whilst groups of weanling rats were fed a high-carbohydrate fat-free diet for 2 weeks, and the expected elevated levels of fatty acid synthetase and stearoyl-CoA desaturase activities were achieved, when the animals were switched to the same diet supplemented with 5% (w/w) corn oil containing 60% (w/w) linoleic acid, then both enzyme activities decayed, with a half-life of 2-3 days for fatty acid synthetase and < 12 h for the stearoyl-CoA desaturase (Fig. 10). This observation clearly identified that the nutritional control of hepatic lipid synthesis cannot be by the action of linoleic acid on fatty acid synthesis but rather via its action on desaturation since the time course of its action fell within the diurnal feeding pattern of the rat [83,84].
Jeffcoat, R., & James, A. T. (1984). The regulation of desaturation and elongation of fatty acids in mammals.
So if we go back to “Low fat: Basically a high MUFA diet” we see that DNL is basically an alchemical process that transforms carbs into something(fat) that can be stored seemingly indefinitely, and the local SCD1 is activated to deal with the excess saturation in places where DNL can be very active. And here it seems to indicate that suffering from an “unsaturation deficiency” causes SCD1 to enslave DNL in order to obtain its raw material.