EFA deficiency, Mead Acid and other endogenous PUFAs!
-
This is just a post about my curiosity (and possibly that of others here) about EFA deficiency, Mead Acid production and how a living being copes with a deficiency of exogenous PUFAs. As I said in another post, I tend to write a lot because I like the subject, so I've divided it into topics for those who want to go into something specific.
- Delta-6 desaturase and PUFAs: Mead Acid is always produced, just not a lot
- What changes occur in EFA deficiency that increase Mead Acid production so much?
- Endogenous PUFAs: Mead Acid is not the only one!
Delta-6 desaturase and PUFAs: Mead Acid is always produced, just not a lot
Mead Acid is an omega-9 20:3, produced from the desaturation of oleic acid 18:1, and receives the most attention in studies on EFA deficiency. The truth is that we produce it every day, it's not exclusive to omega-3/6 deficiency.
I see it in a similar way to how they use the term “ketosis”, everyone produces/metabolizes ketones all the time, “ketosis” is just the term used to indicate a greater presence of them. I found it funny when they joked about the term peatosis and mead acid being a peatone in X.
D6D acts on 3 main fatty acids (this is what you'll see in most articles), Alpha-Linolenic(18:3n-3)>Linoleic(18:2n-6)>Oleic(18:1n-9), this is also the order of preference defined by delta-6 desaturase. Mead Acid is produced all the time, what happens is that the desaturation of Oleic is strongly suppressed by the presence of ALA and LA. These 3 compete with each other and preference and quantity influence the decision as to which inhibits the other.
Ullman, D., & Sprecher, H. (1971). An in vitro study of the effects of linoleic, eicosa-8,11,14-trienoic and arachidonic acids on the desaturation of stearic, oleic and eicosa-8,11-dienoic acid.
In the study reported here, the conversion of 18:1 (n-9) to 18:2 (n-9) was a very slow reaction in that a conversion rate of only 2.8% was observed when 10 nmoles of substrate were used. The use of higher levels of substrate did not increase the amount of 18:2 (n-9) produced. Although both 18:2 (n-6) and 20: 3 (n-6) did inhibit the conversion of 18:1 (n-9) to 18:2 (n-9) the 20:4 (n-6) did not significantly alter the desaturation rate of 18:1 (n-9). In these studies on the inhibition of 18:1 (n-9) desaturation it should be noted that we are measuring a low rate of conversion and are not necessarily using a saturating level of 18:1 (n-9).
Enzymatic studies have also shown that 18:3 (n-3) is desaturated more rapidly than 18:2 (n-6) which in turn is a better substrate for desaturation than is 18:1 (n-9). Enzymatic studies have also established that 18:3 (n-3) as well as 18:2 (n-6) effectively inhibit the conversion of 18:1 (n-9) to 18:2 (n-9). Although the number of desaturase enzymes in the microsome is not known, it has been suggested that a common enzyme may act on 18:1 (n-9), 18:2 (n-6) and 18:3 (n-3) to introduce a double bond at the 6-position.
GURR, M. I. (1974). The Biosynthesis of Unsaturated Fatty Acids. Biochemistry of Lipids, 181–235.
Only unsaturated fatty acids of similar chain length significantly depressed the conversion of oleic, linoleic and linolenic acids into more unsaturated acids. The depressive effect increased with unsaturation in the order 18:3>18:2>18:1. These results correlate well with results in vivo in respect to the inhibition of 5,8,11-20:3(Mead Acid) synthesis by fat deficient rats after supplementation of the diet with linoleic or linolenic acid.
What changes occur in EFA deficiency that increase Mead Acid production so much?
I think it's right to say that the conversion of Oleic to 18:2n-9 (and subsequently Mead Acid) is defined by a ratio of (ALA+LA) to Oleic, the higher this ratio in favor of ALA+LA the lower the Oleic desaturation. Since it's hard to find someone who gorges on ALA, LA is the main suppressor of Mead Acid by preference and quantity, so removing it in studies results in Mead Acid production.
So Mead Acid is produced all the time, but its levels (as far as desaturase is concerned) are defined by this ratio. I'm going to use the levels presented in one study, obviously it doesn't reflect the reality of how it works because you have other mechanisms at work so it's just to understand the general “timeline” of what happens for D6D to start desaturating Oleic. At least that's how I see it.
Brenner, R. R., Garda, H., De Gómez Dumm, I. N. T., & Pezzano, H. (1981). Early effects of EFA deficiency on the structure and enzymatic activity of rat liver microsomes.
0 days of EFA-deficient diet:
- LA: 12.8
- Oleic: 5.6
- LA to Oleic ratio: 2.2
- Mead Acid: 0
4 days of EFA-deficient diet:
- LA: 10.1
- Oleic: 9.7
- LA to Oleic ratio: 1. 04
- Mead Acid: 0
11 days of EFA-deficient diet:
- LA: 4.1
- Oleic: 12.8
- LA to Oleic ratio: 0.320
- Mead Acid: 1.1
23 days of EFA-deficient diet:
- LA: 5.1
- Oleic: 17.4
- LA to Oleic ratio: 0.293
- Mead Acid: 4.9
So when you pass a certain threshold, the lower the ratio, the more the Oleic can be desaturated and the omega-9 PUFAs are no longer “undetectable”. When a fatty acid composition test reports that your Mead to Arachidonic ratio is greater than 0.4, you are classified as having an “essential fatty acid deficiency”.
Honestly, I don't know what the ratios would look like, if you consider D6D's preferences maybe something like ((ALA3)+(LA2))/Oleic
Endogenous PUFAs: Mead Acid is not the only one!
Mead Acid receives almost exclusive attention in EFA deficiency studies, and I think this special attention it receives leads one to think (at least I was led to! haha) that endogenous PUFAs are only formed from omega-9, and since Mead Acid is not the substrate for Cyclooxygenase then we also come to the conclusion that there are no prostaglandins in a state of total EFA deficiency. So much to uncover!
First of all, in the absence of exogenous PUFAs, desaturases can act on other fatty acids that are not limited to omega-3/6 or omega-9 (ironically, the old studies are the only ones I found that addressed this lol):
Wolff, R. L., Sebedio, J.-L., & Grandgirard, A. (1990). Separation of 20∶4n−6 and 20∶4n−7 by capillary gas-liquid chromatography.
Several investigators (1-11) have observed that fat deficient diets can lead to the appearance of polyunsaturated fatty acids of the n-7 series in phospholipids. These acids include 18:2Δ8,11 , 18:3Δ5,8,11, 20:2Δ10,13, 20:3Δ7,10,13 and 20:4Δ4,7,10,13 acids.
Schmitz, B., Murawski, U., Pflüger, M., & Egge, H. (1977). Positional isomers of unsaturated fatty acids in rat liver lipids.
In a similar way, 9-16:1 seems to become available for several metabolic conversions normally occupied by the fatty acids of the (n-6) family. The sequence 6,9-16:2; 8,11-18:2; 10,13-20:2 as well as 6,9-18:2; 8,11-20:2 are in accordance with the findings of Bernert and Sprecher (14). Besides desaturation of 9-16:1 to 6,9-16:2, chain elongation yields 11-18:1. This acid may then serve as a substrate for further desaturation as indicated by 6,11-18:2; 5,11-18:2; and 7,13-20:2. These results show that cis-vaccenic acid (11-18:1) is subjected to several metabolic reactions in rat liver during essential fatty acid deficiency.
A number of fatty acids listed in Table II do not fit into any of the four fatty acid families. Among these, the odd numbered fatty acid 5,8,11-19:3 may be produced by the reaction sequence 9-17:1 -> 6,9-17:2 -> 8,11-19:2 -> 5,8,11-19:3. Other even and odd numbered monoenoic acids not belonging to any of the families may be the products of a-oxidation. Four of the octadecadienoic acids listed in Table II are not contained in Scheme II. The (n-5) fatty acids may be genetically linked by the following reaction sequence 9-14:1 -> 11-16:1 -> 13-18:1 -> 9,13-18:2. A similar Δ9-desaturation of 12-18:1 has been postulated in essential fatty acid deficient rats (7), although no such desaturation could be found by other workers in the field (8,32). 6,12-18:2 is probably formed by Δ6-desaturation of 12-18:1. This monoenoic fatty acid could, however, not be detected in the present study although its occurrence in mammalian tissue is well documented (33-35). Similar pathways have to be postulated for 4,7-18:2 and 5,8-18:2 (36)
Eight different fatty acids were identified with double bonds in position 5: 5,8-18:2; 5,11-18:2; 5,11-20:2; 5,8,11-18:3; 5,8,11-19:3; 5,8,11-20:3; 5,11,14-20:3; and 5,8,11,14-20:4. They all posess at least one double bond in the same position as the genuine substrate of the Δ5-desaturase system at C-atom 8,11, or 14 with chain length between 18 and 20 C-atoms. A Δ4-desaturase system is postulated for the conversion of 7,10,13,16-22:4 into docosapentaenoic acid: 4,7,10,13,16-22:5 (37). The identification of 4,7,10,13-20:4 belonging to the (n-7) family shows that in fat deficient rats 7,10,13-20:3 is also accepted as a substrate.
Now with regard to prostaglandins, it seems that we can't say that they don't occur without omega-3/6 either. There are some studies with different fatty acids that have been synthesized and it seems that having a certain structure and the double bonds in certain positions, prostaglandin isomers can be created (so the question is whether the potency is similar).
Here the fatty acid 7,10,13-20:3 n-7 produces an isomer of PGE1. Perhaps the 4,7,10,13-20:4 found in EFA deficiency will also become a PGE isomer if the double bound at position 4 doesn't render it unusable as a substrate.
Struijk, C. B., Beerthuis, R. K., Pabon, H. J. J., & van Dorp, D. A. (2010). Specificity in the enzymic conversion of polyunsaturated fatty acids into prostaglandins.
The acid found by Mead in EFA-deficient animals, 20: 3 n9, did not show any conversion at all. On the other hand, 20:3 n7, also 'found to a less extent in EFA-deficient animals, can be converted into PGE1 isomer.
This isomer, however, does not show any biological activity in the guinea pig ileum test, neither do the other unnatural prostaglandins
I'm not saying that these isomers of PUFAs and prostaglandins fulfill the same function, just that there are several unexplored possibilities. It sounds interesting and at least contrary to some of the claims out there.
-
@TexugoDoMel
“D6D acts on 3 main fatty acids, preferably in this order: ALA (18:3n-3) > AL (18:2n-6)> Oleic (18:1n-9)”.
=> Thanks for posting and let us remind we can manage thanks to mead acid (W9) when we haven’t taken EFA for several days, though we can’t yet say mead acid role is going to drive to prostaglandins. Other nutrients come into account.
I would like to highlight too that D5D is the first limiting step in converting EFA to prostaglandins. Δ5-desaturase has a particular higher affinity for ALA (W3), provided we do not overeat PUFAs.
So when eating white fish with fried food (fried with peanut or sunflower oil), we won’t make much PGE3. Omega-6 and omega-3 fatty acids share the same metabolic pathways. Excessive intake of omega-6s can interfere with the conversion of omega-3s to beneficial compounds like PGE3.Competition for Δ5-desaturase
Both ALA and LA can be substrates for Δ5-desaturase. However, when ALA is abundant and LA intake is not excessive, Δ5-desaturase tends to favor ALA, leading to increased production of omega-3 LC-PUFAs. In typical western diet the ratio W6/W3 is 15/1, and even 20/1 in USA (corn and soy).
Figure to help some readers: Enzyme pathway for linoleic acid (W6) and useful nutrients for conversion
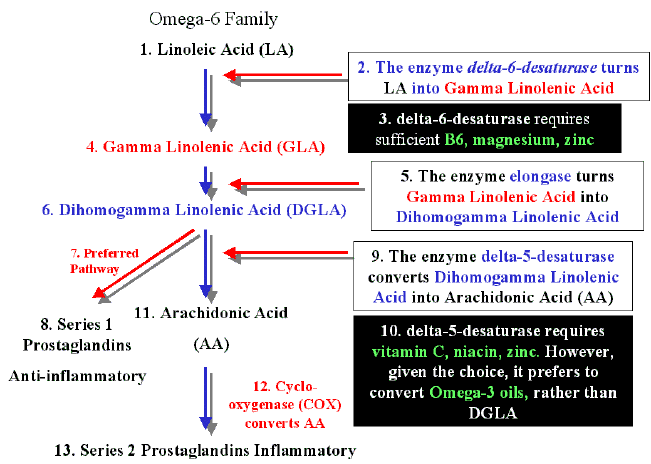
Sources:
- Omega−3 Polyunsaturated Fatty Acids (PUFAs): Emerging Plant and Microbial Sources, Oxidative Stability, Bioavailability, and Health Benefits—A Review.
doi: 10.3390/antiox10101627 2021 NIH.
Excerpt:
The highest conversion of ALA to EPA occurs when LA and ALA are supplied at the ratio of 1:1. Our capacity / facility to convert depends on our genes. The activity of Δ6- and Δ5-desaturase enzymes is largely modulated by variants in encoding genes (FADS1-2-3 gene cluster) and determines the bioconversion rate of ALA to VLC-FUPAs.
The capacity to convert pant-based LC-PUFAs into VLC-PUFAs, is dominant in Americans (59%), common in East Asians (57%) and Europeans (35%), and largely absent in South Asians (14%), and Africans (2%) [47].
If Asian and African population has little access to seafood, there will be a rather low rate of ALA to VLC-PUFAs [47].
- Inhibitory Factors for D6D Enzymes. Principles of Orthomolecularism. R.A.S. HEMAT. Urology and health. 2004. P 346-348: & OBESITY (from books.google.com)
Excerpt:
The activity of D6D can be blocked by a host of factors: 1- trans-fatty acids (common in hydrogenated oils, margarines and shortenings), 2- high saturated fat intake, 3- cholesterol, 4- deficiencies of zinc, pyridoxine (vitamin B6), or magnesium, 5- diabetes (i.e., severe insulin deficiency), 6- excessive alcohol intake, 7- aging, 8- oncogenic viruses, 9- chemical carcinogens and 10- ionizing radiation.
Avoiding hydrogenated oil/margarine-based food products; eating only low-fat meat, poultry and dairy products; minimizing alcohol intake; avoiding chemical additive-containing processed/manufactured (junk) foods; and taking supplements of zinc (15 mg/day), vitamin B6 (10-50 mg/day) and magnesium (200-500 mg/day), will tend to maximize D6D activity, at least somewhat increasing conversion of cLA to GLA. Vitamin B6 may also aid the conversion of GLA to DGLA for conversion to cAMP – enhancing PGE1. Vitamin C and niacin (vitamin B3) are needed to convert DGLA to PGE1; so supplements of vitamin C (300-500 mg/day, minimum) and vitamin B3 (50-100 mg/day) may also aid PGE1 formation.
Supplements of preformed GLA from evening primrose oil, borage oil or blackcurrant oil may be helpful. DGLA can be converted to AA by the enzyme D5D. The primary activator of D5D is insulin. The primary hormonal suppressor of D5D is glucagon, and the fish-oil fatty acid EPA (eicosapentaenoic acid) is also a significant inhibitor of D5D. Feedlot beef, etc., is rich in AA; low-fat range-fed beef, poultry, etc., is low in AA, and contains some EPA. Growth hormone and insulin are both anabolic. Growth hormone promotes fat burning/loss, while insulin opposes fat burning and promotes fat gain. Increased insulin levels and decreased growth hormone levels arc characteristic of obesity. PGE1 suppresses insulin release; PGE1 increases pituitary growth hormone release. Growth hormone-releasing hormone requires adequate pituitary cAMP levels to perform its growth hormone-releasing. Lowering insulin through a low-carbohydrate diet combined with GLA/EPA supplements to enhance PGE1/cAMP levels may restore age-declining GH function. Growth hormone helps to build muscle mass when combined with testosterone. Obesity/high carbohydrate diet-elevated insulin inhibits the testosterone-producing activity of FSH/LH. In both men and women, testosterone may be converted to estrogen through an aromatase enzyme. The aromatase enzyme exists and functions primarily in body fat. Estrogen is a powerful pro-fat hormone, deposits in the fat of the breasts, subcutaneous tissues, the buttocks and thighs. Insulin, estrogen and cortisol are the 3 primary pro-fat hormones of the human body. Severe chronic stress is a threat to normal male testosterone levels. Both testosterone and cortisol are made from the precursor prohormone pregnenolone. Normal daily mate testosterone production is 5 mg, while 10-20 mg of cortisol is produced daily under non-stressed life conditions. The amount of cortisol produced under stress may double, stealing scarce pregnenolone needed for (decreasing with age) testosterone production. Cortisol is extremely pro-fat, and is the chief agent of muscle catabolism. The late twentieth century Western epidemic of obesity is as much due to widespread chronic hypokinesis (too little bodily movement), as it is to the carbohydrate/caloric excess typical of modem humans. Exercise decreases storage fat rather than LBM (lean body mass), whereas dietary interventions, i.e., dieting, tend to reduce both body fat and LBM. Exercise increases insulin sensitivity and decreases insulin resistance.
- Omega−3 Polyunsaturated Fatty Acids (PUFAs): Emerging Plant and Microbial Sources, Oxidative Stability, Bioavailability, and Health Benefits—A Review.
-
Thanks for the info!
There are some animal studies on restricting only omega-6, and the effect is to increase the metabolism of omega-3 and decrease that of omega-6:
Fifteen weeks of n-6 PUFA deprivation compared with adequate diet decreased the mean unesterified plasma concentration of n-6 PUFAs by 84% (Table 1). The change in the total unesterified n-3 PUFA concentration was statistically insignificant, although unesterified DHA and EPA concentrations were increased by 53% and 79%
The total n-3 PUFA concentration in brain was increased by 15%, reflecting largely an 11% increased DHA concentration (Igarashi et al. 2009). Expression of enzymes of the 20:4n-6 cascade, cPLA2-IVA and COX-2, was downregulated, whereas expression of DHA-preferring calcium independent iPLA2-VIA (Garcia and Kim 1997; Strokin et al. 2004; Ramadan et al. 2010; Basselin et al. 2011) and of 15-lipoxygenase (LOX) was up-regulated (Kim et al. 2011).
COX-2 mRNA was decreased significantly (−23%, p b0.05) (Fig. 3A) in the n-6 PUFA deprived rats compared with adequate rats, as was COX-2 protein (−32%, pb0.05) (Fig. 3B). The deficient diet did not change COX-1 or mRNA significantly