Aspirin metabolism: glycine and beyond
-
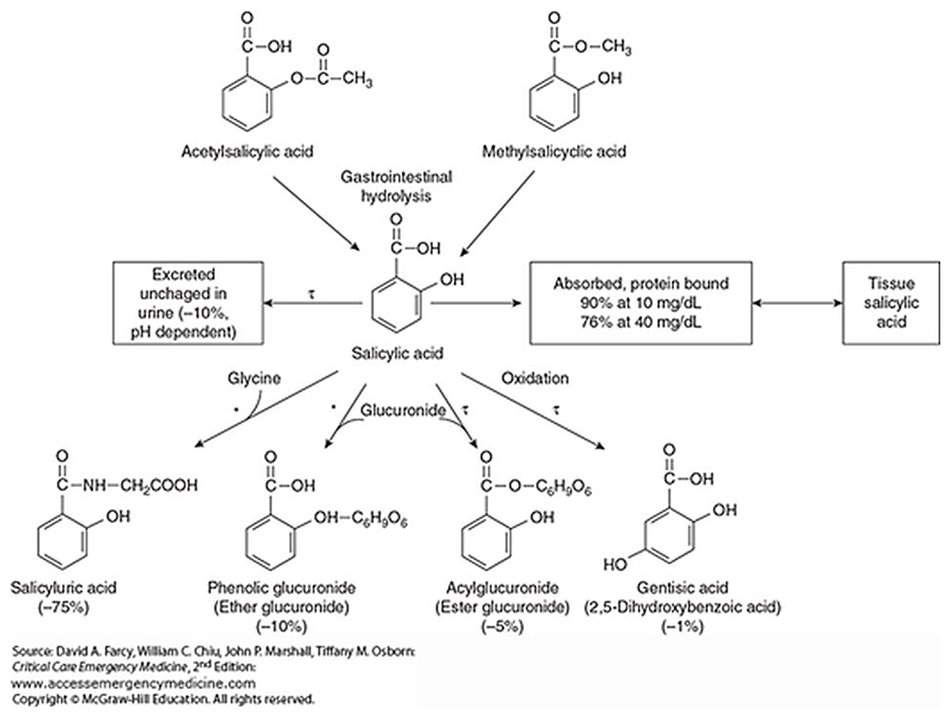
Metabolism of Aspirin after Therapeutic and Toxic Doses
"Caldwell et al.[28] reported a large inter-subject variation in the urinary recovery of salicyluric acid (range 5.9-71.6%, some 12-fold variation) and salicyl glucuronides (0.1-90%) in 85 healthy volunteers who took 900 mg ASA by mouth. Another recent report from the same laboratory[8] again showed that the urinary excretion of SA and metabolites in volunteers who ingested ASA 900 mg was highly variable. Such larger inter-individual variation was not observed in the present study (
Figure 1).""The present work also explores the kinetics of SUA formation from SA under conditions of increasing aspirin load from therapeutic to toxic amounts. In the three groups of subjects from whom the mean values of total salicylate recovered were 246, 2999 and 8095 mg, the proportions comprising SUA were 75, 47 and 22% and those comprising SA were 9, 31 and 65%, respectively. Thus the effect of saturation of SUA formation is clearly demonstrated at doses that extend well into the toxic range, although we recognize that urine alkalinization would have influenced the pattern in the most severely poisoned group. Reduced excretion of salicylate as SUA in those that took overdoses of aspirin was accompanied by increased elimination as GA and SPG indicating that the processes that lead to the production of these metabolites continue to play a significant part (22-33%) in the inactivation of salicylate."
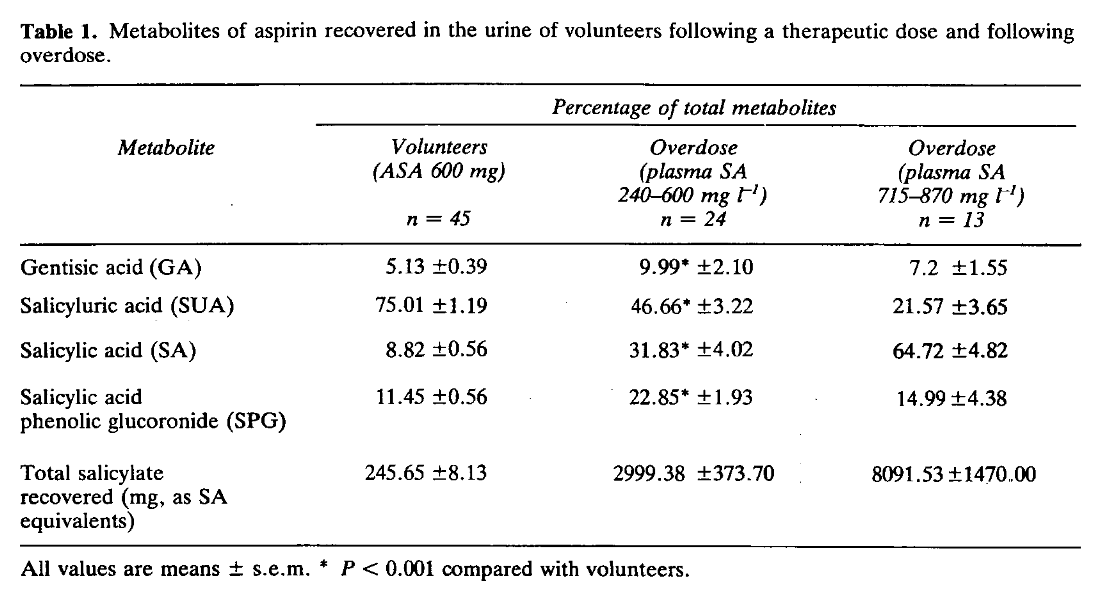
Since not all of the aspirin dose is conjugated to glycine, we may simplify as (85%):
- 0.85 glycine (75.07 g/mol) per 1 aspirin (180.16 mg/mmol).
Along with discounting the glycine-unrelated excretion, we have to adjust for mass differences:
- 75.07 g/mol ÷ 180.16 g/mol ≈ 0.4
Therefore..
- 0.85 (glycine conjugation fraction) × 0.4 (mass correction factor) ≈ 0.3
ASA has 3 letters, making it easy to remember the 30% for the corresponding glycine depletion.
If a victim takes 500 mg of aspirin, this dose will deplete only (500 mg × 0.3 = ) 150 mg of glycine.
From there, the higher the dose, the lower the fraction of glycine conjugation. Based on Table 1:
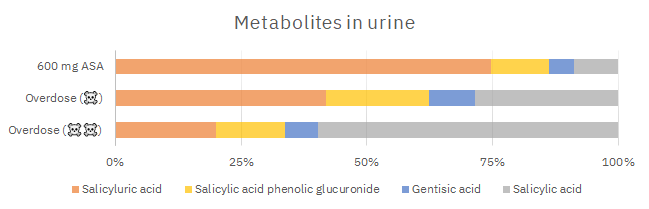
For the two 'overdose' groups of the report, it would be about 20% and 10% with the previous considerations. Suppose that they took 10 g of aspirin (those 8 g of SA equivalents), such dose would deplete 1 g of glycine.
Pharmacokinetics of aspirin: evaluating shortcomings in the literature
"Even though aspirin is one of the most widely used drugs, the literature on the detoxification of salicylic acid by the major glycine conjugation route is severely limited. Salicylic acid is detoxified via a two-step enzymatic process in the mitochondria of the liver (Figure 1).
- The xenobiotic/medium chain fatty acid:CoA ligase (ACSM2B, EC 6.2.1.2) uses ATP to activate salicylic acid to salicyl-CoA releasing AMP and pyrophosphate [70].
- Glycine N-acyltransferase (GLYAT, EC 2.3.1.13) forms salicyluric acid and releases CoA by conjugating salicyl-CoA to glycine [71]."
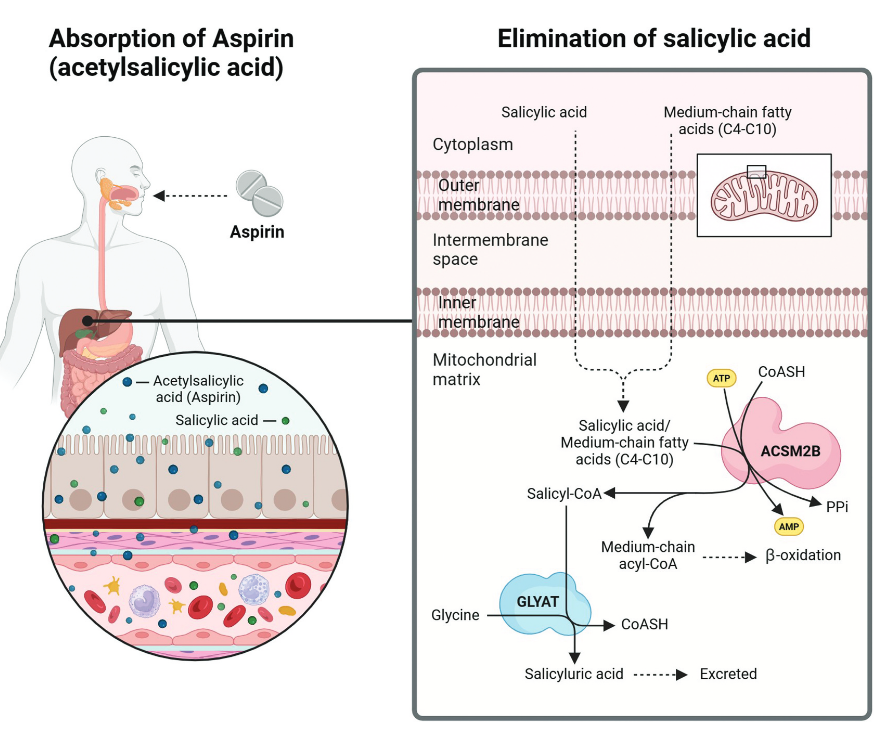
"The first enzyme involved in the pathway, namely ACSM2B, can form salicyl-CoA at a low efficiency (<1%); however, studies on bovine ACSM2B have shown an inhibitory effect of salicylate. The ACSM2 ligases and their role in xenobiotic metabolism is understudied, particularly the effect of genetic variation on enzyme activity; the identity of factors affecting ACSM ligase activity in vivo, the role of inhibitors on the conjugation ability of ACSM2B and the association between ACSM2B and detrimental health outcomes."
"Glycine conjugation is saturable and can, therefore, only detoxify a limited range and concentration of substrates. The rate of glycine conjugation can be affected by the formation of the acyl-CoA (ACSM2B) and/or the conjugation of the acyl-CoA to glycine (GLYAT)."
"The rate of glycine conjugation can be affected by:
- The available co-factors (ATP, CoA, and glycine);
- Genetic variants in the ACSM2B and GLYAT genes;
- Different expression levels of ACSM2B and GLYAT;
- Disease states.
Studies have shown that although ATP can limit glycine conjugation of salicylate, glycine concentration is not a limiting factor."
"The availability of glycine may be a limiting factor in glycine conjugation of benzoate [94,98,99]. It can influence the rate of detoxification as was demonstrated by the dose dependent increase in benzoylglycine (hippurate) formation after administration of glycine [94]. Benzoylglycine is formed from benzoate and is the preferred substrate for detoxification by the glycine conjugation pathway. However, even though the formation of salicylate has been found to be capacity-limited at therapeutic doses [69], oral administration of glycine does not increase the excretion of salicyluric acid. This indicates that glycine is not the limiting co-factor. However, the available amount of ATP for the reaction can be reduced by salicylate itself as it uncouples oxidation and phosphorylation [65]. This then limits the rate of the reaction."
"In a study investigating the effect of orally administered glycine on salicylate metabolism during aspirin overdose, it was found that the plasma glycine was lower in the overdose patients suggesting depletion of available glycine. Orally administered glycine increased the plasma glycine concentration while the fraction of total salicylate recovered as salicyluric acid remained the same. This supported previous studies, however, orally administered glycine did shorten the time it took to reach the maximum rate of salicyluric acid excretion [100]."
"The glycine conjugation pathway is often oversimplified, as it's not just limited to detoxifying benzoate into hippurate. This pathway plays a crucial role in metabolizing and detoxifying various substrates, including natural compounds from food like salicylate, dietary polyphenols, and medium-chain fatty acids (MCFAs), as well as xenobiotics and metabolites from organic acidemias [98,101,157,158] (Figure 2). Xenobiotics, such as salicylate and MCFAs, are first activated to acyl-CoA. Salicyl-CoA is conjugated to glycine, while MCFA acyl-CoAs are further processed by β-oxidation [159]."
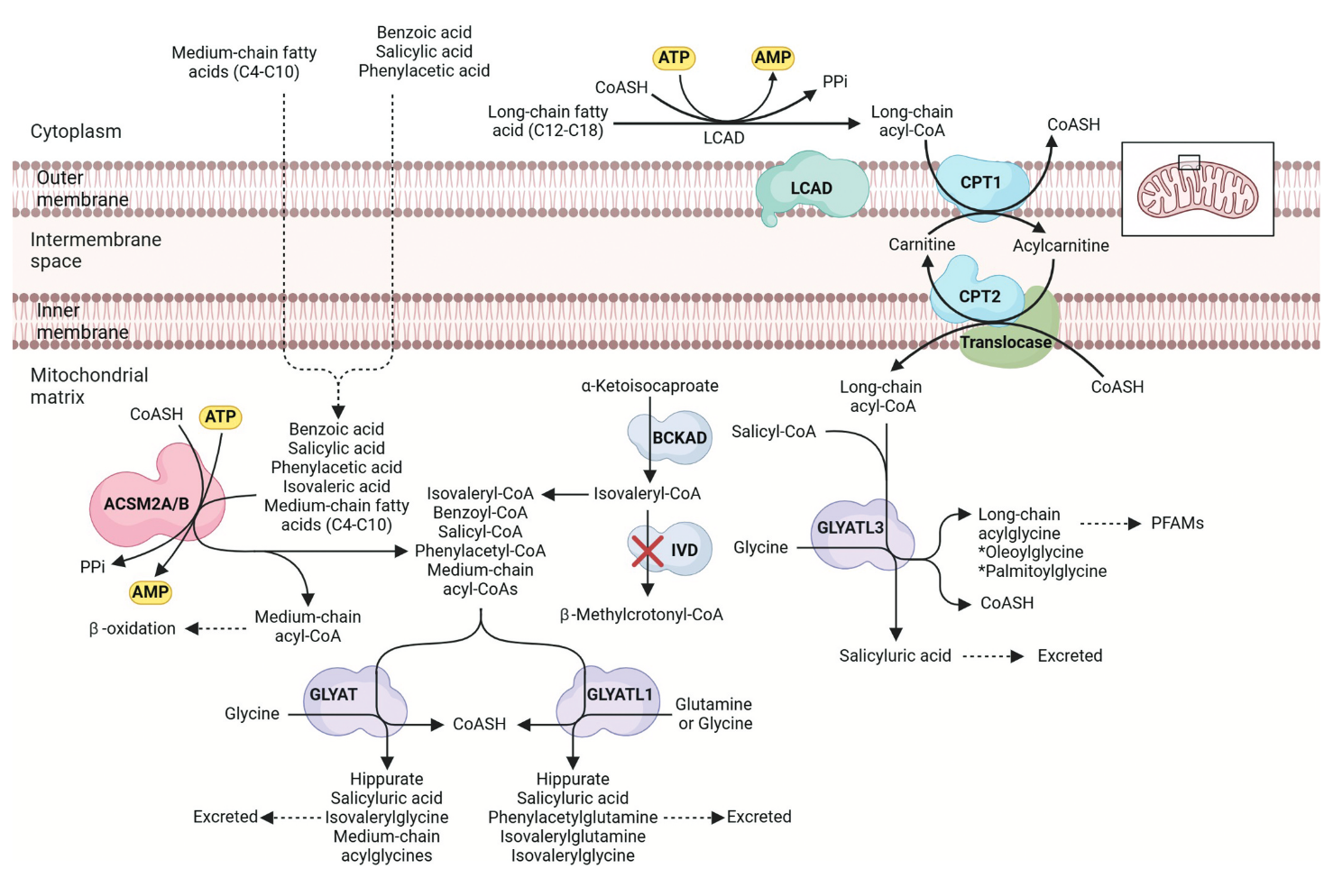
"The glycine conjugation pathway evolved to metabolize endogenous substrates (e.g. MCFAs), and therefore, xenobiotics such as salicylate add an additional detoxification load. It has not been determined if these multiple substrates have a deleterious effect on the rate of glycine conjugation, but it is known that the pathway can be saturated [64,92,93,97,109,162,163]. This probably indicates that the preferred substrate will be detoxified at a rate that does not exceed a threshold concentration and that less preferred substrates will accumulate."
"[..]salicylate first needs to be activated to a salicyl-CoA before it can be conjugated to glycine to form the glycine conjugate (Figure 2). Previous studies have shown that the activation step occurs at a much slower rate than the glycine conjugation rate [69,74,154,155]. This is because salicylate is not the preferred substrate for ACSM2B [70]."
"[..]the formation of salicyluric acid reaches a maximum rate at higher doses of aspirin (1 g or more) [153]."
"Furthermore, benzoate, a preservative found in many food items, can outcompete salicylate for glycine conjugation, increasing the half-life of salicylate. Microorganisms in the gut produce benzoylCoA from dietary polyphenols. This contributes a very large load that needs to be metabolized by the glycine conjugation pathway [161]."
"Humans are consuming and increasing concentration of substrates that need to be detoxified by the glycine conjugation pathway. These include natural sources (e.g. salicylate found in berries [158]), polyphenols [196], and xenobiotics [197,198]. Therefore, the ability of an individual to detoxify salicylate will also depend on the type of diet they follow, as a diet high in preservatives (benzoate) will reduce the amount of salicyluric acid that is formed. Salicyluric acid formation can be inhibited for long periods if even small amounts of benzoate are repeatedly ingested [199]."
"Salicylates are [..] found in many common food items, e.g. vegetables, fruits, herbs/spices, and beverages [35,36]. Significantly higher levels of salicylic acid were found in the serum of vegetarians not taking aspirin, with the reported levels comparable to that of patients taking 75 mg of aspirin [37]."
"The metabolism of salicylic acid in cancer patients are severely limited [153] and it has been shown that the GLYAT gene is transcriptionally downregulated in hepatocellular carcinoma [118,203]. Cancer cells have a high demand for glycine and serine for growth [204]. The inhibition of glycine uptake or biosynthesis can impair cancer cell growth [123]. Therefore, the GLYAT gene is downregulated in cancer cells to prevent the enzyme from depleting hepatic glycine, which would inhibit the rapid proliferation of cancer cells [118,123,124,205,206]. It is possible that aspirin may exert its anticarcinogenic properties via the induction of expression of GLYAT which then leads to the depletion of hepatic glycine and inhibition of cancer cell growth. It has been shown that the long-term use of salicylate may increase the production of salyciluric acid by induction [97,207,208]."
-
Pharmacokinetics of Salicylate in Man
"Only the nonionic form of salicylic acid is subject to tubular reabsorption. As a consequence, the renal clearance of salicylate is exquisitely sensitive to urine pH. As urine pH is increased from 5 to 8, the renal clearance of salicylate increases more than twenty-fold [39]. While this has a relatively small effect on the elimination kinetics of small single doses (≤0.65 g aspirin in adults), it has a profound effect on the elimination of larger doses, i.e., when the formation rates of salicyluric acid and salicyl phenolic glucuronide approach saturation. This is particularly apparent when urine pH is increased from below 6 to 7 or higher. Consequently, alkalinization of urine is a commonly employed measure for treating salicylate intoxication."
"The alkalinizing effect of sodium bicarbonate on urine pH is well known. Less widely appreciated is the fact that certain "nonsystemic" antacids can also increase urine pH. The term "nonsystemic" is therefore a misnomer. Such widely used antacids as aluminum and magnesium hydroxide and calcium carbonate-glycine mixture can increase urine pH significantly [40]. The lower the baseline urine pH, the greater is the effect of the antacid [40]. Since antacids are frequently taken concomitantly with aspirin (often without knowledge of the physician) to reduce or prevent aspirin-induced dyspepsia, an awareness of this potential interaction is important."
-
@Amazoniac said in Aspirin metabolism: glycine and beyond:
Pharmacokinetics of Salicylate in Man
"The alkalinizing effect of sodium bicarbonate on urine pH is well known. Less widely appreciated is the fact that certain "nonsystemic" antacids can also increase urine pH. The term "nonsystemic" is therefore a misnomer. Such widely used antacids as aluminum and magnesium hydroxide and calcium carbonate-glycine mixture can increase urine pH significantly [40]. The lower the baseline urine pH, the greater is the effect of the antacid [40]. Since antacids are frequently taken concomitantly with aspirin (often without knowledge of the physician) to reduce or prevent aspirin-induced dyspepsia, an awareness of this potential interaction is important."
Is this implying that it is actually better to take aspirin without a "buffer" like sodium bicarbonate? Edit: I read it wrong, this actually states the opposite correct?
-
Yes, alkalinization tends to improve excretion of acids that ionize. The unionized forms are prone to leak from the kidney filtrate and lead to unwanted reabsorption.
What I find confusing in overdosing aspirin is that the most abundant metabolite appears to be salicylate, but it only protonates to salicylic acid when the pH is too low for the typical of urine, let alone for kidneys' tubules.
The pH role in the transport of active principles of drugs through agitated bulk liquid membrane
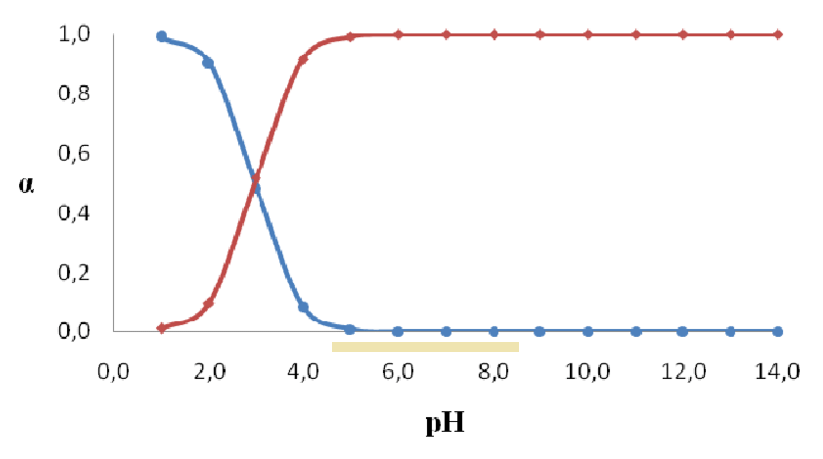
Therefore, the principle above don't seem to apply, unless the urine is extremely acidic, a shy fraction gets reabsorbed, and it becomes significant over time.
-
What does all this mean as it relates to Aspirin and Glycine consumption? K.I.S.S
-
Not much glycine is lost with usual aspirin doses (30% or less of the aspirin weight).
The amount of sodium eliminated in pairing with salicylate ions should also be low. Approximations:
Aspirin
- 1 mmol per 180 mg (from 180 g/mol⇈)
- 5.5 mmol per 1000 mg
Sodium
- 5.5 mmol of sodium is 130 mg (from: 5.5 mmol × 23 mg/mmol)
Maximum direct sodium loss:
- 130 mg Na/g aspirin
It may seem negligible, but it depends on the amount consumed of each.
For a person ingesting an average amount of sodium (4 g), taking high doses of aspirin can induce the depletion of a considerable share of the sodium intake.
As for the alkalinization, it may enhance the activity of acyl-CoA synthetases (ACS), mentioned above as the limiting step, similar to the disinhibition effect on some glycolysis enzymes.
-
A few issues concerning the salicylic acid analogue mentioned in this recent article.
Salicylic acid
- 2-hydroxybenzoic acid (2-HBA)
γ-Resorcylic acid
- 2,6-dihydroxybenzoic acid (2,6-DHBA)
Variations in the additional hydroxyl group relative to salicylic acid (orange):
Hydroxybenzoic acid isomers and the cardiovascular system
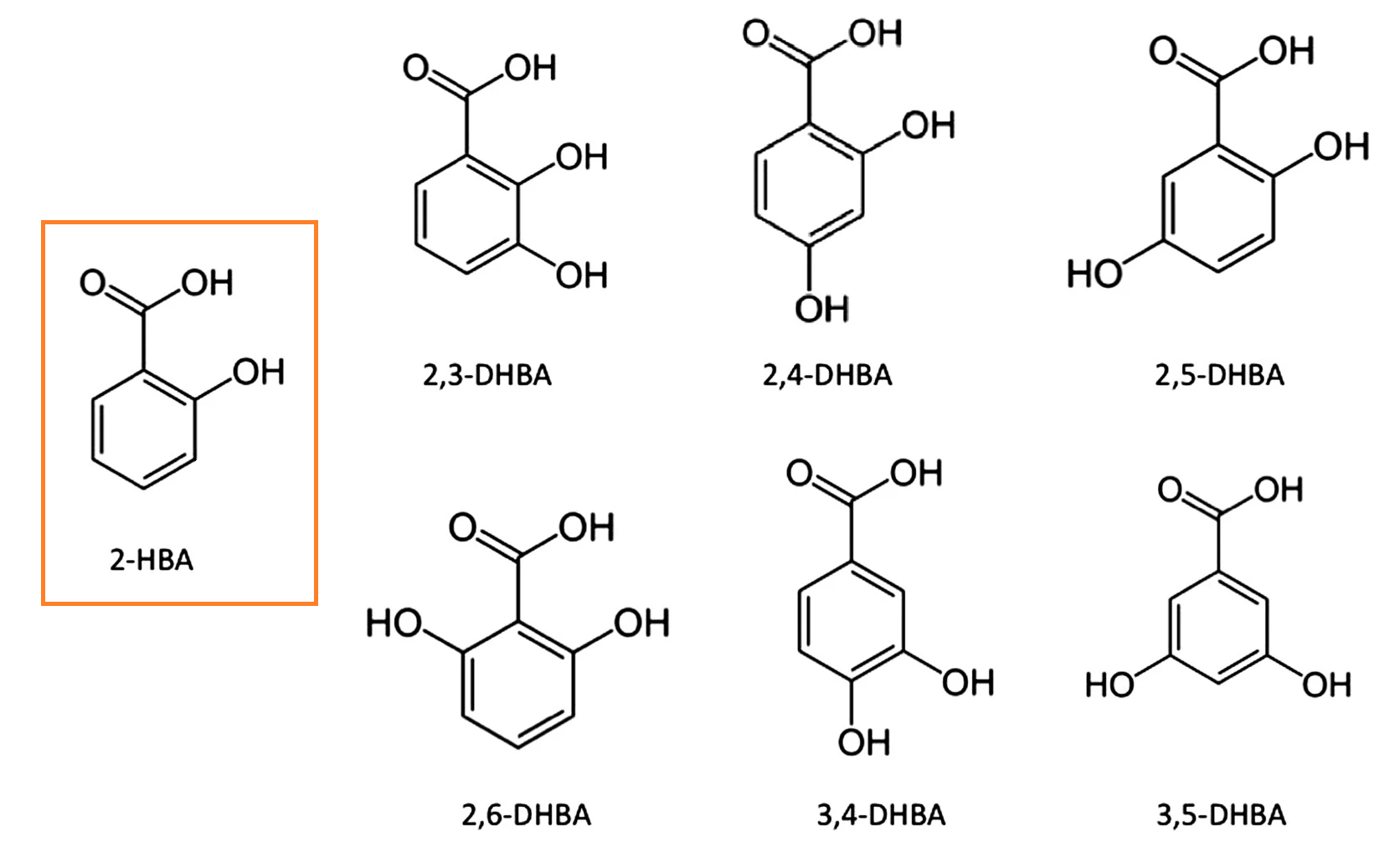
The coordination in 2,6-DHBA helps to stabilize the molecule in an ionized state:
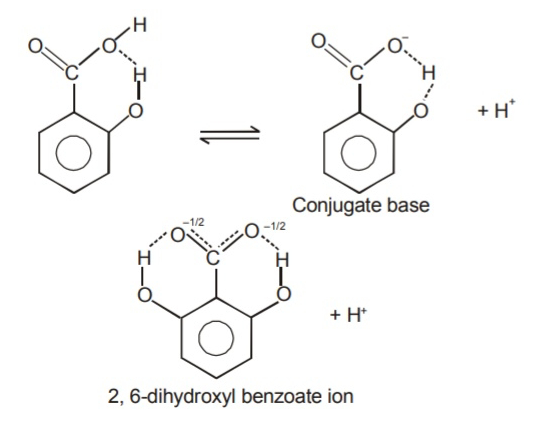
Source: the internet.I think that this property is responsible for its lower pKa, making it a 'stronger' acid, that's more prone to deprotonate (benzoic acid → benzoate⁻ + H⁺) and stay as such.
pKa:
- 2-HBA: 3.0
- 2,6-DHBA: 1.3
This does make 2,6-DHBA a molecule with greater acidification potential. However, the pH in the body is much higher than needed to ionize them, making the difference irrelevant:
- 50 mmol 2-HBA → 50 H⁺ mmol
- 50 mmol 2,6-DHBA → 50 H⁺ mmol
Once we adjust their doses to match for their alleged potencies and possible risk of toxicity, we may get something like:
- 50 mmol 2-HBA → 50 mmol H⁺
- 5 mmol 2,6-DHBA → 5 mmol H⁺
Which casts doubt on the benefit being from relying on a 'stronger' acid.
To counteract the inverted pH of cancer cells, we have to acidify the interior or alkalinize the exterior. 2,6-DHBA deprotonates already in the stomach, which only adds to the burden of extratumoral acidification. In contrast, I just shared with Jennifer a clinic that infuses baking soda in people with cancer.
Ray's suggestion to manipulate acidity through carbonic anhydrases has more basis. The hydrocarbonate ion recirculates in cancer cells and functions as a carrier of protons, taking them from inside, discharging outside, and repeating the process. Inhibition of carbonic anhydrases compromises this removal of protons and creates an unfavorable environment in cells.
Regarding lipophilicity, the greater the degree of hydroxylation, the less lipophilic a molecule tends to be. It's what happens to venom D when it goes from kilciol to kilcidiol and then to kilcitriol.
"The obtained (experimental and theoretical) logP values for HBAs are in the range 0 < logP < 3; this means that these compounds possess from slightly hydrophilic (logP: 0–1) to moderately lipophilic properties (logP: 1–3) [101]. Generally, the disubstituted hydroxybenzoic acids were more lipophilic than the trisubstituted one (gallic acid has hydrophilic properties, and dissolves well in water and other polar solvents). According to the experimental logP values, the compounds can be ordered by increasing lipophilicity as follows: 3,4,5-THB→3,4-DHB~3,5-DHB→ 2,3-DHB→2,4-DHB →2,5-DHB →2,6-DHB."
"Dissociation and association phenomena affect the value of the partition coefficient. Therefore, we observed a relationship between the acid dissociation constants (expressed as pKa) and the values of partition coefficients. Each functional group that can be a hydrogen bond donor or acceptor increases the hydrophilic nature of the compound. Hydroxyl and carboxylic groups present in the structures of the compounds can form hydrogen bonds with water molecules in the aqueous environment, which affects their solubility in water. Therefore, in the case of 3,4,5-trihydroxybenzoic acid, an increase in the hydrophilic properties of the acid is noticeable. The more hydrogen bonds can be formed between a molecule and water molecules, the greater its solubility in water. In the case of ortho- substituted benzoates, the -OH group in the ortho- position is mostly engaged in hydrogen bond formation with the -COOH group, facilitating dissociation of the H+ carboxylic group and increasing the acidity of 2,6-DHB (pKa = 1.30) and, to a lesser extent, 2,3-DHB (pKa = 2.91), 2,5-DHB (pKa = 2.97), and 2,4-DHB (pKa = 3.11). In the case of 3,5-DHB (pKa = 4.04), 3,4-DHB (pKa = 4.26), and 3,4,5-THB (pKa = 4.40), the -OH groups are too far from -COOH to interact with this functional group, but may form hydrogen bonds with the solvent. As a result, the acidity of these acids decreases (higher values of pKa) compared to the ortho-substituted benzoates."
"The dependency between acidity (pKa exp.) and lipophilicity (logPexp.) in the series of studied hydroxybenzoic acids is shown in Figure 7. With the increase in the acidity of the compounds, the lipophilicity increases as well. Therefore, the hydroxybenzoic acids can be divided into three groups characterized by: (a) lower acidity and lipophilicity (3,4,5-THB; 3,4-DHB; 3,5-DHB), (b) moderate acidity and lipophilicity (2,3-DHB; 2,4-DHB; 2,5-DHB), and (c) higher acidity and lipophilicity (2,6-DHB)."
That's counterintuitive.
But here's the value for salicylic acid:
"Experimental n-octanol-water partition coefficients (logPexp) determined by traditional shake-flask method were equal to 2.35 and 1.14 for salicylic and acetylsalicylic acids, respectively."
Aspirin is 'rapidly deacetylated' in the body, and salicylate is the main circulating metabolite, which must not differ a lot from 2,6-DHBA in terms of lipophilicity.
Aspirin, stroke and drug-drug interactions
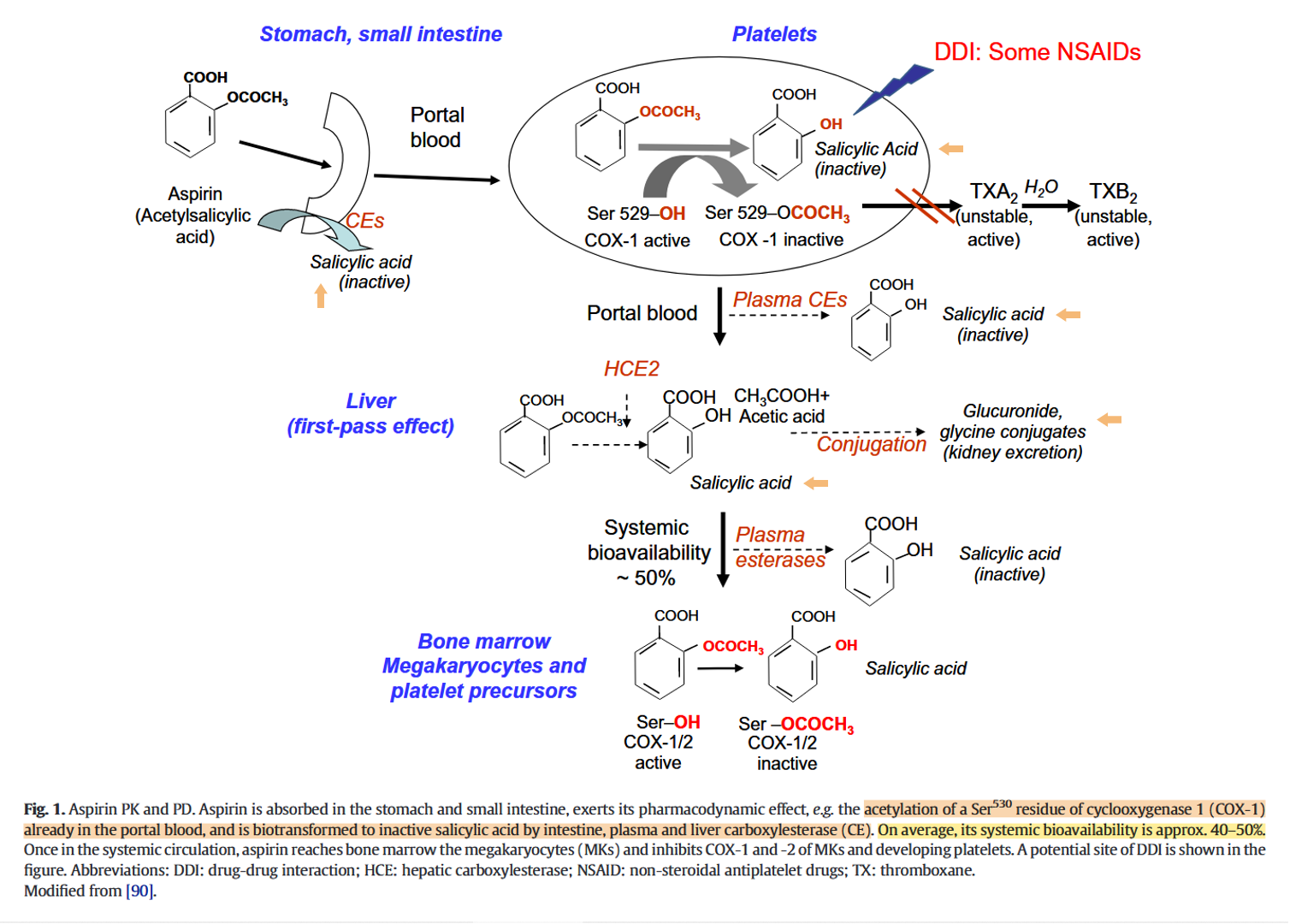
They don't include the small intestine in the first-pass effect, when it's also responsible for the presystemic metabolism of the drug.
Acetyl transference appears to be a neutral reaction. But deacetylation through hydrolysis leads to proton release rather than the expected consumption (to reform the hydroxyl group). A proton from a water molecule is taken up by salicylate and the rest complexes with the freed acetyl group, which occurs deprotonated.
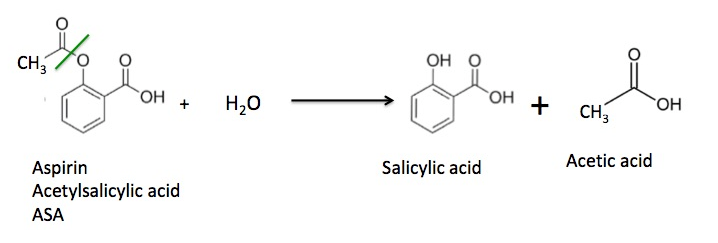
Aspirin and salicylate: An old remedy with a new twist
"Aspirin has a short half-life in circulating blood (~20 minutes) and is rapidly deacetylated and converted to salicylate in vivo. Salicylate does not affect COX-1 or COX-2 activity. Thus, the anti-inflammatory and antineoplastic actions of aspirin and salicylate remain a dilemma. Sodium salicylate paradoxically inhibited prostaglandin synthesis when added to intact cells.[7] Furthermore, healthy subjects taking sodium salicylate excreted a significantly lower amount of prostaglandin metabolites in urine than those not taking sodium salicylate, and their levels of inhibition were comparable to those of patients taking aspirin and indomethacin.[9] Aspirin also reduces human seminal prostaglandin levels.[10] These data suggest that salicylate inhibits COX metabolism by a mechanism different from a direct inhibition of COX activity."
So, part of aspirin won't reach the circulation, deacetylation can occur before systemic distribution, and the other part is soon deacetylated. Salicylate level can remain elevated for hours.
That we will be dealing with ionized forms conflicts with lipophilicity.
Effect of Enteric Coating on Antiplatelet Activity of Low-Dose Aspirin in Healthy Volunteers
"Aspirin is deacetylated to
inactivesalicylate at a number of sites, including the gut; thus, its bioavailability is about 50%. Plain aspirin is absorbed from the stomach, where the low pH protects aspirin against deacetylation and maintains aspirin in a nonionized form which encourages absorption."The pKa of 2,6-DHBA is low enough that it can remain partially ionized even in the acidic pH of the stomach, which prevents intact absorption in an extreme scenario where gastric cancer cells were the desired target.
Therefore, I don't think that 2,6-DHBA being a 'strong' acid is a good reason to adopt it or a convincing explanation for the positive outcomes of use. Aspirin overdose can indeed result in acidosis, but would be an indirect effect.
-
The rationale was: carbon dioxide goes through cells' lipids and releases a proton inside once hydrated. Then, let's find another compound that's also not barred by lipids and releases even more protons. This led to 2,6-DHBA.
It overlooks that the very property that makes the selected compound a greater acidifier is also what makes it extraordinarily prone to deprotonate prematurely. The ability to pass through lipids is likely to be compromised with the early proton release. If it could be injected into the tumor, it would still be uncertain if it could be taken up by target cells.
Healthy Cancerous Extracellular pH 7.4 ↓6.5 Intracellular pH 7.1 ↑7.5 For direct internal acidification, it would be more fitting to seek weak rather than strong acids, those that can take advantage of the bump in transitioning from the extracellular to intracellular compartment (6.5
 7.5) to deprotonate the molecule where intended.
7.5) to deprotonate the molecule where intended.
Hello! It looks like you're interested in this conversation, but you don't have an account yet.
Getting fed up of having to scroll through the same posts each visit? When you register for an account, you'll always come back to exactly where you were before, and choose to be notified of new replies (either via email, or push notification). You'll also be able to save bookmarks and upvote posts to show your appreciation to other community members.
With your input, this post could be even better 💗
Register Login