A combination of vitamin B1/B3/B7 and aspirin, has curative effects on human mantle-cell lymphoma
-
@OxidisedOolong yep, weight is holding steady, I’ve also noticed that I don’t seem to need the upper end of the calories anymore, (I’ve had to really be cognizant of whether I’m eliciting a stress reaction or not, and eat accordingly). I’ve settled more into the 3800 calorie side of the equation, (maybe the additional ATP let my body upregulate an immune response temporarily and I just needed more calories to get over that hump?).
-
@evan-hinkle Cool, that's great to hear - solid evidence that it raised your metabolic rate.
-
Just a quick update:
I have a sensation of much more room in my throat, (in the past swallowing has been difficult, and it’s always felt very tight in my throat due to the nodules).Clicking that I used to experience when swallowing has gone away, ( I assume again this had to do with a lack of room in the throat to perform swallowing correctly).
In general I feel as though the experience has been positive, and I’m not sure I would attribute any negative effects to it thus far. In the interest of being transparent, (after being a terrible sleeper for many years) I have become, since finding Peat’s work, a great sleeper. Since embarking on the B vitamins and aspirin regimen, I am finding that I’m waking maybe once a night, (sometimes not) and this is new for me having slept through the night for about three years now. Typically I remedy this with a little ice cream or an 8oz glass of milk. Just wanted to mention it since it seems to be related. I’m also regularly, but not nightly, taking .5 mg of cypro to help with the sleep issues.
I’ll be three months in at the end of April.
-
@evan-hinkle how’s it going bud? Any further improvements or positive things you’ve noticed?
-
@haidut said in A combination of vitamin B1/B3/B7 and aspirin, has curative effects on human mantle-cell lymphoma:
Yay, my first post here!
Wanted it to be about the recent study I did. While the results will have to be replicated with a follow-up experiment, with more animals, and additional groups for comparison, this results has never been seen before with this tumor type, which is 100% lethal and has no known case of spontaneous regression (i.e. disappearing on its own, without treatment). Basically, 2 out of 3 animals were fully cured in this experiment/study.
https://twitter.com/haidut/status/1751716166387597730Human-equivalent doses for the substances were ~15mg/kg vitamin B1 (thiamine Hcl), 30mg/kg vitamin B3 (niacinamide), 1.5mg/kg vitamin B7 (biotin), and 15mg/kg aspirin. Administration was once daily, orally.
Good for those who can handle a B vitamin complex !
I can't, whether metylated or not, it makes me anxious, nervous and depressed.
-
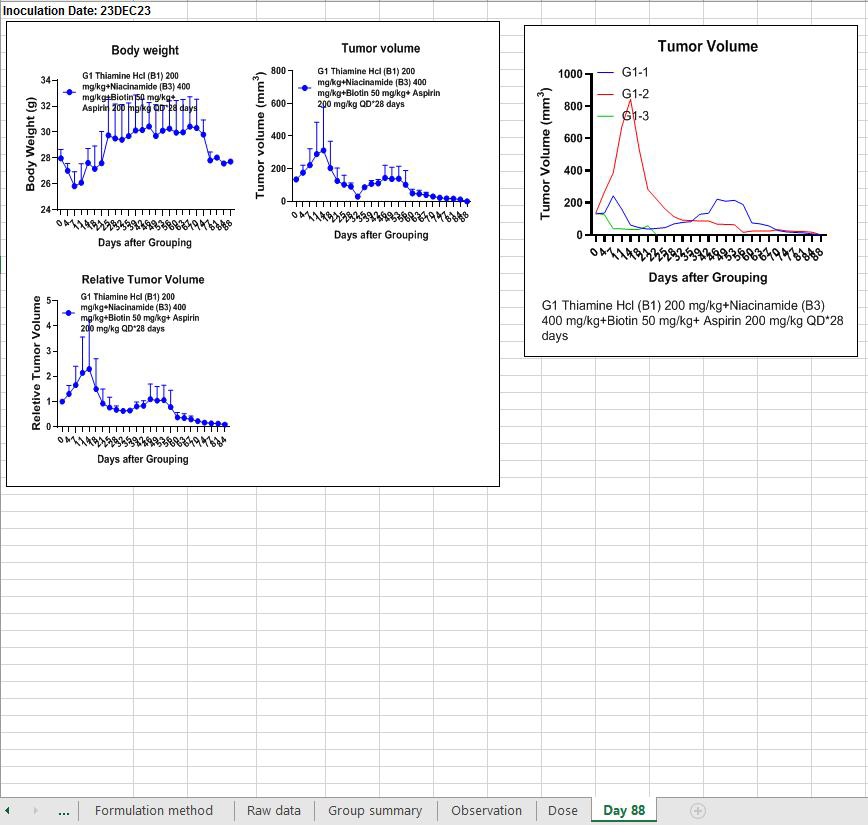
All 3 mice are now fully cured and cancer-free.
I dont know why Georgi isn't making a bigger deal out of this. Because this is a big deal.
https://x.com/haidut/status/1758122576411279659
https://x.com/haidut/status/1777337478636007790
-
@Santosh said in A combination of vitamin B1/B3/B7 and aspirin, has curative effects on human mantle-cell lymphoma:
Good for those who can handle a B vitamin complex !
I can't, whether metylated or not, it makes me anxious, nervous and depressed.
I have the same exact problem. I can handle them once every 1-2 weeks, but if I take B-vitamins on a daily basis I quickly get bonked out.
-
@Mauritio said in A combination of vitamin B1/B3/B7 and aspirin, has curative effects on human mantle-cell lymphoma:
All 3 mice are now fully cured and cancer-free.
I dont know why Georgi isn't making a bigger deal out of this. Because this is a big deal.
I am assuming that he is waiting for an offfical academic publication. In the past he published with a term of people who may be working in a 'publish or perish" environment.
The information that he released on twitter started a 1-year clock ticking. It is sometimes referred to as a 'patentability clock'. He has only 1 year to file a US patent application for a method of treatment and a unique combination of materials for treatment. If he missed the deadline for filing, his disclosures on twitter (older than 1 year) can be used against him to render his ideas "old" and obvious.
-
@evan-hinkle You wrote:
I'll be 3 months in at the end of April.I wonder when we can say "we are cured" when following this kind of supplement plan. I wouldn't want to do this forever. Is 30 days enough? How about Evan's 90 days?
There has to be an end to it otherwise it's no cure at all.
-
@cremes just as an update, I still have my thyroid nodule. I was taking the combination in the morning after breakfast and I don’t think I was well fed enough for the powerful combination. I switched to after lunch and am tolerating it much better. That said, seemingly no change in my nodule, som somewhat bummed, but am willing to continue with the protocol at lunch where I have a better base on food on board.
-
Couple of general thoughts. The B’s were mixed into the chow of the rats. I wonder if they grazed on it all day or if they ate in one sitting? Grazing would have could dole the dose out at a drop vs a one shot.
I’m curious to see the results of the aspirin substitute that Georgi is testing. Apparently it lowered the regression curve from three weeks to one week.
Also, I believe the B’s and aspirin are no longer needed after tumors are gone. I may have misheard, but during a recent podcast with Mercola, Georgi mentioned that the tumors did not return, (and I thought he said this was after ceasing the B’s/aspirin)? Maybe someone else can confirm?
-
@evan-hinkle said in A combination of vitamin B1/B3/B7 and aspirin, has curative effects on human mantle-cell lymphoma:
@cremes just as an update, I still have my thyroid nodule. I was taking the combination in the morning after breakfast and I don’t think I was well fed enough for the powerful combination. I switched to after lunch and am tolerating it much better. That said, seemingly no change in my nodule, som somewhat bummed, but am willing to continue with the protocol at lunch where I have a better base on food on board.
Thanks for the update, I appreciate the details.
-
@Crypt-Keeper have you tried taking a lower dose? Perhaps splitting the capsule into 5 capsules? Hence a lower RDI % etc.
-
Georgi speaks about the result of his study here.
-
@DavidPS thanks for posting this, it answered many of my questions.
Sure enough, treatment was stopped after full regression, and the tumors did not come back.
-
It might be that he is waiting for a publication, but Im not sure if he really wants to patent this treatment. Did he say that anywhere ?
Plus, I'm not sure if you can patent a combination of basic vitamins. Normally you would have to change something structurally on the molecules ...Dare to think.
My X:
x.com/Metabolicmonstr -
@Mauritio - Yes, it appears that he is not interested in patenting.
He selected the least expensive form of B1 to make his invention accessible to as many people as possible.
I am a registered patent attorney. I think he could obtain a patent based on the limited facts that I have. He solved a long-standing problem; that is a strong indication of patentability.
A mind is like a parachute. It doesn’t work if it is not open. 👀
☂️ -
I’m continuing to plow through this interview. Georgi is now running a follow up study with three groups of rats including the vitamins and aspirin in one group, and another group with ONLY 2,6 dihydroxybenzoic acid, (no B vitamins ).While the tumors are regressing as expected in the aspirin vitamin group, the 2,6 dihydroxybenzoic acid group is already cured!
https://www.sigmaaldrich.com/US/en/product/aldrich/d109606
I am not a doctor, this is not medical advice. The above link is for research purposes only.
-
@evan-hinkle said in A combination of vitamin B1/B3/B7 and aspirin, has curative effects on human mantle-cell lymphoma:
Between this and some vitamin K3 we could lose weight and cure our metabolic diseases at the same time.
")
BTW, I'm serious about the latter part. As bioenergeticists, we believe that most/all diseases are brought about by broken or sub-optimal metabolism. If it's true, then this protocol should also serve to potentially solve type2 diabetes, heart disease, hypertension, IBS, dementia, alzheimers, OBESITY, and autoimmune diseases like MLS, Lupus, etc.
I have heart disease. I'm going to try this protocol and see what happens. I'm already taking B1, low-dose B3, and aspirin daily. I just need to adjust the amounts a bit and add in some biotin.
-
Anyone watched Georgi’s interview on the strong sistas channel? He goes into discussing his study in quite some depth:
Hello! It looks like you're interested in this conversation, but you don't have an account yet.
Getting fed up of having to scroll through the same posts each visit? When you register for an account, you'll always come back to exactly where you were before, and choose to be notified of new replies (either via email, or push notification). You'll also be able to save bookmarks and upvote posts to show your appreciation to other community members.
With your input, this post could be even better 💗
Register Login