Vitamin D Receptor stops mitochondria respiration [Why vit D can cause problems] [1,25 vitamin D]
-
This is gonna be a long ass post on vitamin D mechanisms and how while its needed, it can be harmful in certain situations involving low ATP (not just calcium) as it can be an anti-metabolic substance, and reasoning for why it could cause the issues people often report
showing
- Vitamin D, in its more potent 1,25 form, slows metabolism / ATP & co2 production. as a function (sometimes) necessary to some extent
- Its through the VDR vitamin D receptor, and likely also through 1,25 vit D binding & antagonizing thyroid receptors
- presence of the VDR (increased by 1,25 vit d) suppresses gene expression effects of the thyroid receptors, and the thyroid receptors suppresses effects of the VDR receptor
- hypothyroid people have higher 1,25 vitamin d, regardless of the level of the usual vit D measure, normal euthyroid people are in the middle, and hyperthyroid people have lower 1,25 vitamin D than both. due to excess conversion to the 1,25 form
- a way to fix issues with this could be by raising t3 thyroid hormone / fixing hypothyroidism, ensuring enough iron intake (balanced), and / or lowering macrophages if there's chronic inflammation.
Some people get side effects from vitamin D, e.g twitching & heartbeat & adrenaline problems, even on 1000iu, ive seen a lot of people on the forum and reddit mention this and have experienced extremes from it, with magnesium making the situation even worse,
there's research that highlights the problems/extremes a subset of people get from taking Vit D are likely due to ATP/co2 depletion from further impairing mitochondria (and maybe not good to take in hypothyroidism).
A few years ago i got into a severe situation from vitamin D where my body was twitching constantly rapidly all over building up for weeks, with sharp heart pain, legs jolting uncontrollably, adrenaline rushes surging me out of sleep with heart racing, and beats skipping & firing all over the place.
Pushing through with magnesium took the situation to a worse extreme & vitamin K didn't do anything. tried different forms of everything and it was the same. So since then i've had to avoid all vitamin D supplements and get not too much sunlight to keep the symptoms mild
Wanted to find out what would cause such an extreme reaction,
twitching is a big sign of ATP depletion / mitochondria problem,
Ray wrote about how when the ATP/Co2 production in the cell is high the cell can shift to a relaxed state.
When the cell is energized, by the mitochondria working with thyroid, oxygen, and sugar, these proteins rapidly change their form, binding calcium and removing it from the contractile system, allowing the cell to relax, to be fully prepared for the next contraction. If the calcium isn't fully and quickly bound, the cell retains extra water and sodium, and isn't able to fully relax.
if you think about this intuitively, when things die & cells have no ATP their bodies tighten up permanently. maybe its whats happening to the body on a micro scale / duration with twitching because of low atp.
Vit D is lowering an already low atp state from hypothyroidism or chronic unresolving inflammation. & the heart is especially vulnerable to low t3 being that it doesnt utilize t4 well, explaining the heart issues with this. & magnesium making it worse is also explained by magnesium being a cofactor needed for the conversion of 25 -> 1,25 vitamin D.So by this, need to fix thyroid before you can tolerate vitamin D well preventing excessive conversion to 1,25 and not having so much around that inhibits an already impaired mitochondria. and also need to lower macrophages if they're elevated (chronic inflammation) as they raise this 1,25 vitamin D form and inhibit mitochondria too.
1st some is clearly needed with key beneficial functions
e.g dopamine functioning, copper import, immunity, wound healing, fixing cognitive disorders, key in schizophrenia, maybe Alzheimers, etc
but the form of it in the body matters for how potent its inhibitory effect on mitochondria can be.Depleting vit D with 0 intake does lower mitochondrial respiration and impair muscles, https://pubmed.ncbi.nlm.nih.gov/33862598/ ,
BUT this is likely an indirect effect or due to effects outside of the VDR by 25 vit d, as i'll show in a bit the DIRECT effect of the vitamin D receptor is to OPPOSE thyroid hormone functioning and IMPAIR mitochondrial energy production (probably supposed to be a "protective" mechanism to lower ROS as well as lower heat production)its function when elevated in its potent 1,25 form (converted from 25 vit d), is to put the brakes on energy production.
(the effects of depletion in muscle there may be due to something secondary, which i think could be copper impairment as vit D is needed for copper to import into cells, which is needed for mitochondria to function)
(the 25 form might be responsible for this with signalling outside of the Vitamin D Receptor, where the 1,25 converted form might do the opposite.)
(it might be due to VDR needing to suppress excess mitochondrial respiration to some extent, so without any of the brakes on paradoxically it gets worse)
,
Here travis discussed how vitamin D lowers heat production, lowers uncoupling protein (maybe as its a signal you are getting heavy sun exposure)
https://raypeatforum.com/community/threads/is-vitamin-d-supplementation-even-neccessary.23844/post-341522Calcitriol has been shown to suppress uncoupling protein, a mitochondrial protein that intimately regulates thermogenesis. This is in fact the proteins classic function, and it had originally been called 'thermogenin'
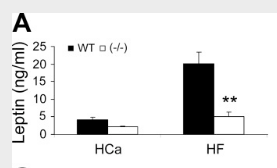
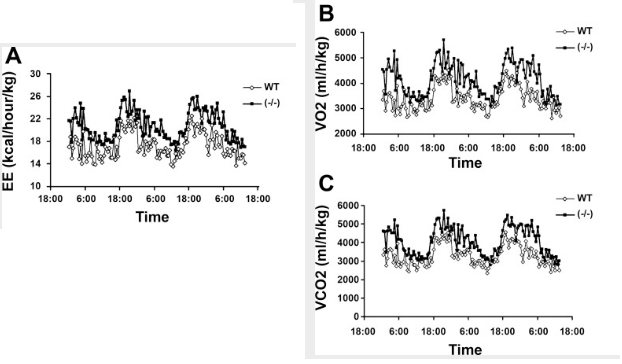
So what effect does this have? The presence of vitamin D₃—acting through its nuclear receptor—suppresses the induction of uncoupling protein, a protein most powerfully upregulated by thyroid hormone and catecholamines.⁽²⁾ This has the effect of lowering metabolism and causing weight gain, something can be demonstrated by using genetically engineered mice**: Mice lacking the vitamin D receptor (VDR) always have a higher metabolic rate**, higher oxygen consumption, and a lower body weight despite consuming the same amount of food as the control mice.⁽³⁾ Conversely, mice having additional vitamin D receptor expressed in their cells invariably have a higher body weight.⁽⁴⁾
And the study there shows if you knock out the VDR in mice, their heat production goes up and they lose bodyfat. Their metabolism increases. aka the VDR is anti metabolic
So after vitamin D₃ is sensed by our cells, our metabolism will be decrease with intent of lowering body temperature. This means that taking additional vitamin D₃ is tantamount to tricking the body into thinking that it's warmer than it really is, and too much of this vitamin–hormone should lower metabolism through this mechanism—by suppressing uncoupling proteins (UCP2 & UCP3)
What exactly is our molecular thermostat if not retinoids and vitamin D₃? I suppose person could imagine a transcription factor in the nucleus serving this role, or perhaps even a receptor somewhere, but I challenge anyone to think of anything besides cis-trans isomerization directing the process.
https://europepmc.org/article/PMC/2670625
confirmed that 1,25-dihydroxyvitamin D3 directly suppressed the expression of the UCPs. Consistently, the energy expenditure, oxygen consumption, and CO2 production in VDR-null mice were markedly higher than in WT mice.
^ uncoupling protein 3 is induced by thyroid hormone, vit D receptor lowers it along with metabolism
so does vitamin D have an opposing effect to thyroid hormone?
-
There are multiple forms of vitamin D. Vit D gets converted to different forms and this conversion can be SKEWED in certain situations
-
1,25 vit D has much more potent signalling than 25 vit D. it takes a lot less to exert effects at the Vit D Receptor. 25 vit D still might have some signalling, and shares most of the same effects on genes as 1,25, but can exert effects outside of the VDR and has a % of gene expression differences
e.g :
The intriguing finding is that the metallothionein 2A gene was found to be a distinct target for 1,25(OH)2D3 but not for 25(OH)D3
- 1,25 vitamin D is often elevated in chronic illness , as conversion from 25 vitamin D is ramped up. e.g macrophages can continually catalyse the conversion to 1,25 without any stopping of the process. magnesium is needed for this.
haidut: "Another important finding of the study is that it was the levels of 25-OH-D (calcifediol, a biomarker of of cholcalciferol (vit. D3) supplementation) and not 1,25-OH-D (calcitriol), which had inverse correlation to all-cause mortality and premature death. "
ray: https://www.youtube.com/watch?v=JOEqOd4ceGA#t=1h44m 1,25 has degenerative effect where 25 has regenerative {but i dont think ray was fully aware of this for his follow up response, in situations where 1,25 is high it means high conversion rate, meaning more 25 will give more 1,25 and make matters worse, e.g 1,25 also went up here after supplements https://core.ac.uk/reader/77027890Unregulated production of 1,25-dihydroxyvitamin D (i.e., sarcoidosis, granulomatous diseases) is an uncommon cause of hypercalcemia;
Vitamin D BINDS THE THYROID RECEPTOR with very high potency in computational model, as an ANTAGONIST,
even more potent than its own receptor- "1,25 vit d3 has, actually, ten times higher affinity for the beta-thyroid, and similarly for the alpha thyroid. That lines up with anecdotal indications we have, that in fact, high levels of 1,25-D in sick people does affect their thyroid function"
"Ultimately, these elevated levels of 1,25-D would displace the endogenous ligands from GR, AR and ThRa. The increasing concentration of 1,25-D would also depress generation of 25-D, leading to a lower concentration of 25-D in serum " (if negative feedback is not functioning well)
^ this also explains something else for me, responding poorly to anti cortisol stuff in hypo (along with the extra reliance on backup energy). its blocking cortisol from exerting its effect too. and maybe why adrenaline raises so high with this problem. {i was waking up in the middle of each night shooting up out of sleep with heart rate beating through the roof}

1,25-D docked into the VDR with a (nanomolar) Kd of 8.48, but also exhibited a Kd of 8.12 into the glucocorticoid receptor (GR), 8.41 into the thyroid-alpha-1 receptor (ThRa) and 8.05 into the androgen receptor (AR) (all Kd values were computed using XSCORE(69,70)). Similar high affinities were found with 25-D, which yielded Kd values of 8.36, 8.17, 8.32 and 8.07, respectively.
Fig. 3A shows the minimum energy (docked) configurations for T3 and 1,25-D in the ThRa, and Fig. 3B for dexamethasone and 1,25-D in the GR. After examining the residues contacted by each ligand, it appears likely that 1,25-D is an antagonist of activation for each of these receptors, but so little is known about the active receptor residues that all one can say **definitively at this point is that the vitamin D metabolites will competitively displace cortisol and T3 from these nuclear receptors **
-
& 1,25 increased in chronic inflammation
1,25-dihydroxyvitamin-D (1,25D) often reaches excessive levels in normocalcemic patients suffering from chronic Th1 inflammatory illnesses, including sarcoidosis and rheumatoid arthritis. This is due to unregulated production of 1,25D in the mitochondria of activated macrophages.
In sarcoidosis, for example, this dysregulated vitamin D conversion can mean that even a moderate intake of vitamin D through ingestion or solar exposure can cause the 1,25D hormone to become high enough to stimulate osteoclastic action, and bone resorption. -
Consequently the ‘‘vitamin D deficiency’’ being observed may well be downregulation of the 25-D metabolite under the influence of the elevated levels of 1,25-D during pregnancy
-
- They joined Barnes et al.(50) in noting that correlation between the 25-D and active 1,25-D metabolites seemed strongest in disease, and weakest in health (because in hypothyroid more is converted to 1,25 so they both go up?)
-
Hypersensitivity to vitamin D occurs because, as part of the Th1 inflammatory response, activated macrophages possess 1-hydroxylase activity and are able to metabolize 1,25D from 25D
-
the inflammation from Th1 activity leads to paracrine extra-renal conversion of inactive 25D to active 1,25D, often depleting serum levels of measured 25D in order to increase active vitamin D (how can It deplete if theres 100x more 25 than 1,25 in plasma?) <←- NOT DIRECT DEPLETION , THE 1,25 EXCESS CAUSES ENZYME ELEVATION THAT METABOLISES 25
-
An angiotensin receptor blocker, olmesartan, was also found to be very helpful through its ability to block angiotensin II from binding to its receptors on the macrophages, thus slowing the increase in activated macrophage production of 1,25D
-
The authors concluded that the elevated levels of active 1,25 vitamin D might be due to inflamed tissue involved in the disease process. This was supported by measurement of elevated levels of 1 alpha hydroxylase (CYP27B1), the enzyme that converts 25D to 1,25D, from colonic biopsies of Crohn’s patients
-
BONE LOSS IN RHEUMATOID ARTHRITIS is lower WITH LOWER 1,25 (hydroxychloriquine can be used to block conversion of 25 → 1,25
In fact, it is interesting to note that of 17 patients with rheumatoid arthritis whom Sambrook et al. [48] found to have bone loss near the wrist joint during a two-year study (mean loss rate 6.1%), one patient actually had a slight increase in bone density near the wrist joint. This patient was also the only one to take hydoxychloroquine during the study period, a drug sometimes used to block the conversion of 25D to 1,25D in sarcoidosis. From examining Table 2 and Fig. 2B [48], one can determine that this patient also had the lowest 1,25D level in the study, and this may account for the lack of periarticular bone loss. Two other patients with almost no bone loss also had very low 1,25D.
The presence of the vitamin D receptor opposes effects of thyroid hormone / the thyroid receptor. when 1,25 vitamin D goes up, thyroid goes down. when 1,25 vitamin D goes up, more VDR is around
studies:(vitamin d increases expression of its own receptor)
- "Vitamin D receptors repress basal transcription and exert dominant negative activity on triiodothyronine-mediated transcriptional activity" https://pubmed.ncbi.nlm.nih.gov/8631908/
suggest a novel repressor function of VDR on T3-mediated transcription which may be significant in tissues where VDR and TR are co-expressed.
- "Thyroid Hormone Receptor Does Not Heterodimerize with the Vitamin D Receptor but Represses Vitamin D Receptor-Mediated Transactivation"
- Hyperthyroid humans = lower 1,25 vit D
- Hypothyroid humans = higher 1,25 vit D
(both regardless of normal vit D measures) https://academic.oup.com/jcem/article-abstract/51/4/793/2678274?redirectedFrom=fulltext&login=false
28 hyper vs 42 normal vs 73 hypo
The concentration of 25-hydroxyvitamin D3 was not altered, but the concentration of 1,25-dihydroxyvitamin D3 was significantly lower in the serum of hyperthyroid patients (28 ± 11 ng/liter) than in the serum of normal subjects (42 ± 13 ng/liter). On the contrary, an increased concentration of 1,25-dihy-droxyvitamin D3 was observed in the serum of hypothyroid patients (73 ± 28 ng/liter; P < 0.001 vs. normal subjects).
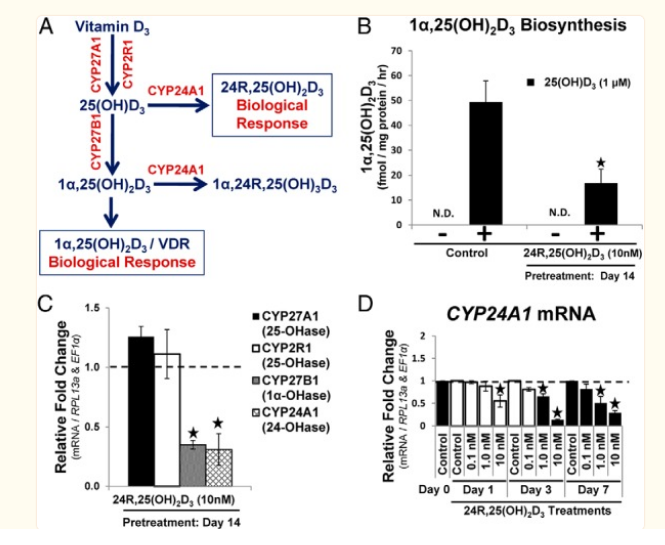
- 24-hydroxylase (CYP24A1), is responsible for the catabolism of 25(OH)D3
The Vitamin D receptor (VDR) works counter to the thyroid receptor
Hypothyroid = elevated conversion to 1,25 vit D (higher 1,25 levels than normal thyroid, regardless of similar Vit D levels)
Hyperthyroid = lower conversion to 1,25 vit D (lower 1,25 levels)
- low calcium diet (~150mg) makes it worse with added conversion to 1,25 form to increase intracellular calcium more. (but with 1,25 staying elevated outside of renal conversion, maybe more sensitive to dietary calcium also? assuming it isnt enough to lower the 1,25 enough, because of the lack of t3 action in hypothyroidism, or continued inflammation).
More insight into mechanisms:
1,25 is supposed to re-stimulate t3 production, and regulate itself
. Because 1,25(OH)2D3 enhances TSH-induced thyrotropin secretion (43), T3-dependent suppression of plasma 1,25(OH)2D, mediated by regulation of renal CYP27B1 gene expression, implies the existence of a novel negative feedback pathway along the hypothalamus-pituitary-thyroid gland axis.
High Levels of Active 1,25-Dihydroxyvitamin D Despite Low Levels of the 25-Hydroxyvitamin D Precursor - Implications of Dysregulated Vitamin D for Diagnosis and Treatment of Chronic Disease
Our data show that active1,25D hormone may be elevated, even with a low level of 25D substrate because of the inflamed macrophages’ hyperactive conversion to the active hormone.
Data presented here suggest that this extra-renal synthesis of 1,25D is more widespread than previously thought and because it leads to vitamin D hypersensitivity
://pubmed.ncbi.nlm.nih.gov/3113918/
high dose of T3 thyroid hormone downregulates the enzyme that increases 25 conversion to 1,25 vit D (CYP27B1) and lowers 1,25 vitamin D levels
A megadose of 20mcg/100g so ~2mg heq lowers plasma active vit D acutely in 24hrs
(guess lower dose for longer would have same effect , going by hyper patients?)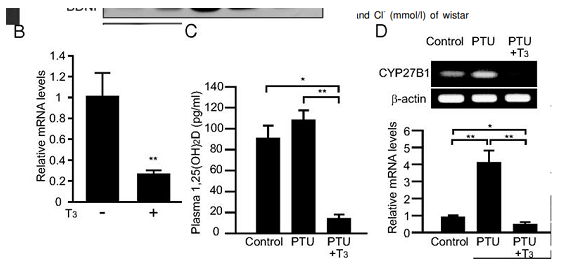
Intervention study showing giving T3 to rats lowers 1,25 vit D,
Making them hypothyroid increases 1,25 vit Dhttps://academic.oup.com/endo/article/154/2/609/2423282?searchresult=1
also increases on low calcium / phosphate diet
- Renal CYP27B1 mRNA levels were significantly increased in groups fed a low-Pi (0.02%) or low-Ca (0.02%) diet when compared with mice fed the control (0.6% Pi and Ca) diet
low calcium diet 0.02% (~150mg heq vs 4.5g calcium) , 2x higher active vit D and massive increase in activating enzyme mRNA
t3 = also decreases 24,hydroxylase(CYP24A1) which is what metabolises d3 but to -50%, where the activator is decreased -80%. so decreases both the activator and the metaboliser (guess makes sense as less active vit D around so dont want it metabolised as much), but the net effect is lower 1,25 active vit d. hypo increases metaboliser 8x (more activate vit D around to metabolise) as well as increasing the activator 4x. but net effect higher 1,25
Intriguingly, coadministration of a low-Ca diet and T3 induced hypocalcemia <--- Need good calcium in diet for t3? (as calcium absorption lowered with less 1,25)
in vivo data showed that transfection of TR, in fact, repressed VDR-mediated transcription and that the repression could be reversed by the addition of RXR. {slightly?} Thus, in vitro and in vivo experiments do not support ligand-sensitive transactivation mediated by VDR-TR heterodimer formation but rather suggest that TR expression can repress 1,25-(OH)2D3-induced transcription predominantly by sequestering RXR
Basically taking vitamin D in a hypothyroid state makes hypothyroid symptoms worse.
and at extremes a lot worsehttps://www.reddit.com/r/Hypothyroidism/comments/bokeee/leg_muscle_twitching_when_undermedicated/
Why would magnesium make this problem so much worse?
[i.e laying in bed twitches rippling all over my body continuously, heart all over the place pretty sure it was gonna give out any moment, jolting out of sleep with adrenaline rushes] ,
probably because magnesium is involved with increasing 1,25 vit D.
when u lower intake of magnesium 1,25 drops.
helping more 25 -> 1,25 vit D converting enzyme to function with more magnesium thats already upregulated high by being in a hypothyroid state, which has an inhibiting action on mitochondria, probably the reasonSilencing the Vit D receptor = increases mitochondria respiration, across all cell types tested
Vitamin D Receptor Is Necessary for Mitochondrial Function and Cell HealthVitamin D receptor (VDR) mediates many genomic and non-genomic effects of vitamin D. Recently, the mitochondrial effects of vitamin D have been characterized in many cell types. In this article, we investigated the importance of VDR not only in mitochondrial activity and integrity but also in cell health.
The silencing of the receptor in different healthy, non-transformed, and cancer cells initially decreased cell growth and modulated the cell cycle. We demonstrated that, in silenced cells, the increased respiratory activity was associated with elevated reactive oxygen species (ROS) production. In the long run, the absence of the receptor caused impairment of mitochondrial integrity and, finally, cell death. Our data reveal that VDR plays a central role in protecting cells from excessive respiration and production of ROS that leads to cell damage. Because we confirmed our observations in different models of both normal and cancer cells, we conclude that VDR is essential for the health of human tissues.SO ^ the major role of the VDR is to put the brakes on mitochondria, ordinarily helping it in terms of not ramping up too much.
Due to the increased ROS that comes as a natural process of mitochondria activity, without the vitamin D receptor putting the brakes on the mitochondria gets damaged and the cell dies. And also redirecting some stuff for cell growth instead.
But if excessive or excess conversion or already in a defective mitochondrial state i.e hypothyroidism, with more u get too much impairment.https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4278832/
We have previously demonstrated that VDR controls mitochondrial respiratory activity We found that transcription of the subunits II and IV of cytochrome c oxidase was significantly increased upon VDR silencing. Accordingly, treatment of HaCaT cells with vitamin D downregulated both subunits, suggesting that VDR may inhibit the respiratory chain and redirect TCA intermediates toward biosynthesis. we examined various acetyl-CoA-dependent biosynthetic pathways, such as the mevalonate pathway (measured as cholesterol biosynthesis and prenylation of small GTPases), and histone acetylation levels; all of these pathways were inhibited by VDR silencing.
VDR knock down in HaCaT cells nearly abolishes VDR expression and drastically reduces cell growth.
On the basis of our observations, we concluded that, in all cell types analyzed, VDR was an essential negative modulator of mitochondrial respiration, and its ablation increased both the expression and the activity of the respiratory chain, and the consequent ROS production.These data provide evidence of the role of VDR as a gatekeeper of mitochondrial respiratory chain activity and a facilitator of the diversion of acetyl-CoA from the energy-producing TCA cycle toward biosynthetic pathways that are essential for cellular proliferation.
Based on our observations, we conclude that the VDR, by restraining mitochondrial respiratory activity, allows the cell to spare metabolic intermediates, which may be diverted from oxidative metabolism toward a biosynthetic fate, supporting cell growth. We validated the general role of the VDR as an enhancer of cellular proliferation extending our observations to several human cancer cell linesBased on these observations, we concluded that the VDR inhibited the mitochondrial membrane potential and likely restrained ROS production, protecting the cell from additional oxidative stress. On the contrary, VDR loss increased the respiratory potential, but rendered cells more prone to an oxidant-driven potential collapse. This possibility was supported by the significantly lower glutathione (GSH) consumption in wild type cells, as revealed by the higher levels of the antioxidant molecule that were measured in wild type cells compared to silenced cells
VDR silencing decreased the de novo synthesis of cholesterol. We demonstrated the exemplary diversion of acetyl-CoA, the incorporation of which is reduced during cholesterol biosynthesis, prenylation events and histone remodeling.
The human proliferating keratinocyte cell line HaCaT does not respond to the antiproliferative action of vitamin D (S1 fig.), and we previously demonstrated that nuclear translocation of the VDR, which is a prerequisite for transcriptional activity, is not induced upon ligand stimulation [12], thus indicating ineffective, or feeble, nuclear VDR signaling in these cells. Therefore, HaCaT cells represent a good model that can be used to examine the mitochondrial effects of VDR activity in a background were the differentiating properties of vitamin D have been lost.
We had evidence that the VDR balances electron chain activity, resulting in dual advantages for the cell: protection from oxidative stress and support for proliferation through readily available biosynthetic intermediates
We concluded that the VDR, by restraining mitochondrial respiratory activity, spares mitochondrial metabolic intermediates, which can be diverted from oxidative metabolism toward a biosynthetic fate. The end products that were found to be affected by this switch in the present study are essential for proliferation; in particular, cholesterol,-
acetyl-CoA that is produced in the cytosol is a crucial component of several biosynthetic pathways that promote cell growth, and our study describes the VDR as a promoter of acetyl-CoA utilization outside of the mitochondria for the first time.
-
Quiescent cells primarily utilize the TCA cycle to oxidize nutrients, generating NADH and FADH2 to fuel ATP production through the mitochondrial electron transport chain, whereas proliferating cells use the TCA cycle to provide the building blocks that are necessary to support cell growth. In this manner, mitochondrial pathways are rewired to sustain proliferation
in our opinion, it is reasonable to conclude that nuclear and mitochondrial VDR signaling are integrated, as described for the glucocorticoid receptor [23], and that further studies will demonstrate both direct and indirect modalities of VDR action on mitochondrial transcription.
1,25 raises intracellular gutathione <---- function is to be dealing with the extra ROS from mitochondria activity?
-
We have previously demonstrated that the specific activity of gamma-glutamyl transpeptidase (gamma-GT), an enzyme of central significance in GSH metabolism, is regulated in vivo in astrocytes by 1,25-dihydroxyvitamin D3 (1,25-D3)
-
This study demonstrates that both gamma-GT gene expression and specific activity, induced by LPS, are potentiated by 1,25-D3. In contrast, 1,25-D3 does not regulate the expression of other enzymes involved in astrocyte detoxification processes, such as superoxide dismutase or GSH peroxidase. In parallel, 1,25-D3 enhanced intracellular GSH pools and significantly reduced nitrite production induced by LPS. Taken together, these results suggest that gamma-GT, GSH, and 1,25-D3 play a fundamental role in astrocyte detoxification pathways.
-
gamma-Glutamyl transpeptidase (gamma-GT), primarily described as a kidney enzyme, is also expressed in several cell types of the central nervous system (CNS). It is involved in the glutathione cycle and in cysteine transport. Here we report that the specific activity of this enzyme is transiently increased in the rat brain, following a treatment with 1,25-dihydroxyvitamin D3 (1,25-D3),
-
this positive regulatory effect does not affect endothelial cells of the brain microvessels, but does affect pericytes and parenchymal astrocytes
-
Since gamma-GT is though to participate in the scavenging of reactive oxygen species, these data suggest that 1,25-D3 could be an effector controlling detoxification processes in the brain.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6208166/
https://www.sciencedirect.com/science/article/abs/pii/S0271531705801940
-
Enteral glutathione increased circulating 1,25-(OH)2D3 and decreased 25-OH-D3 in both control and diabetic animals. These results suggest that exogenous glutathione increases 25-OH-D3 1α-hydroxylation both under basal conditions in the normal animal and in diabetes-induced depression.
-
The expression of the VD metabolism genes CYP2R1 and CYP27A1 (25-hydroxylase), CYP27B1 (1-α-hydroxylase), and vitamin D receptor (VDR) were downregulated in the livers of mice fed an HFD (GSH- deficient) compared with control diet-fed group.
-
The expression of CYP24A1 (24-hydroxylase) was significantly increased, which catabolizes both 25(OH)VD3 and 1α,25-hydroxyvitaminD3.
-
HFD-induces GSH deficiency and epigenetically alters VD-biosynthesis pathway genes. This provides a biochemical mechanism for the VD-deficiency and potential benefits of GSH treatment in reducing 25(OH)VD3-deficiency.
A few years ago i got into a severe situation from vitamin D where my body was twitching constantly rapidly all over building up for weeks, with sharp heart pain, legs jolting uncontrollably, adrenaline rushes surging me out of sleep with heart racing, and beats skipping & firing all over the place.
Pushing through with magnesium took the situation to a worse extreme & vitamin K didn't do anything. tried different forms of everything and it was the same. So since then i've had to avoid all vitamin D supplements and not too much sunlight to keep the symptoms mild
Wanted to find out what would cause such an extreme reaction,
twitching is a big sign of ATP depletion / mitochondria problem,
Ray wrote about how when the ATP/Co2 production in the cell is high the cell can shift to a relaxed state.
When the cell is energized, by the mitochondria working with thyroid, oxygen, and sugar, these proteins rapidly change their form, binding calcium and removing it from the contractile system, allowing the cell to relax, to be fully prepared for the next contraction. If the calcium isn't fully and quickly bound, the cell retains extra water and sodium, and isn't able to fully relax.
if u think about this intuitively, when things die & cells have no ATP their bodies tighten up permanently. maybe whats happening to the body on a micro scale / duration with twitching due to low atp.
Vit D is lowering an already low atp state from hypothyroidism. & the heart is especially vulnerable to low t3 being it doesnt work with t4 well, explaining the heart issues with this. & magnesium making it worse is also explained by magnesium being a cofactor needed for the conversion of 25 -> 1,25 vitamin D.So by this u need to fix thyroid before you can tolerate vitamin D well preventing excessive conversion to 1,25 and not having so much around that inhibits an already impaired mitochondria. low iron intake could play a role, many things. and also need to lower macrophages if they're elevated (inflammation) as they raise this 1,25 vitamin D form and so inhibit mitochondria too.
-
I think it's important not to confuse cholecalciferol with calcitriol. They are both called vitamin D, but they have opposing functions in many ways.
Peat and haidut have discussed this many times.And if you look at the positive vitamin D studies : (almost) all cholecalciferol
If you look at the negative ones: calcitriol
What we take as supplements is cholecalciferol . Of course there's conversion, but it's not the same as taking calcitriol .
And if there was a lot of of conversion going on and vitamin D would oppose thyroid I'd find it hard to believe that it does all these things:
Lower cancer, dementia, asthma, all cause mortality, ...and the list goes onhttps://raypeatforum.com/community/tags/vitamin-d
On the muscle twitching issue :
This is my own theory and experience and somewhat bro sciency.
Muscle twitching is low calcium. You should take calcium BEFORE vitamin D (or magnesium or vitamin K) so that when it hits the bloodstream theres already calcium there that vitamin D can use. Vitamin D seems to need calcium and if it's not immediately there it pulls it from the other places ,like the muscles ,which causes cramps or the teeth, which causes demineralization.
Theoretically that shouldn't work as vitamin D doesn't deplete calcium, but practically that is what seems to happening.I used to have issues with taking vitamin D and K and to a lesser degree magnesium. My teeth got really sensitive and after some time I found tiny pieces of tooth in my mouth, which happened only on the days I took vitamin D or K. Now after I started taking calcium before them, teeth sensitivity has gone away and I haven't lost any more teeth.
In my experience Vitamin D certainly does not have an anti thyroid effect. If anything it helps me loose weight ,helps with liver function and feels androgenic.
-
I highly recommend this podcast with peat on vitamin D . It addresses all the issues that come up here and would take too much time to type them out.
An interesting quote from the podcast description:
"High-dose vitamin D without extra calcium supplementation has been associated with increased levels of the active vitamin D metabolite 1, 25(OH)2 vitamin D (calcitriol), and an increase in CTx."
Should available on Spotify too.
-
@Mauritio
there's a high rate of conversion happening in hypothyroidism and chronic inflammation (studies in OP)- vitamin D already converts to 1,25 highly in states of poor health , and 2. (if 25 vit D has some effects that gets around the VDR increase inhibiting thyroid receptors, not increasing VDR that much or something, or like ray mentioned in that audio, receptors act like quick accelerants for effects but arent the whole effect, 1,25 and 25 share a majority of gene expression changes i think >80% in common, but maybe there's enough differences idk) ,
Even if we say it does get around that inhibitory effect in some way, the 1,25 converts from 25, its increase still has an anti thyroid / mitochondria effect and 1,25 is much more potent than 25. likely explaining why taking vit D can cause problems for some ppl in poor health.
a summary is: if someone has any of: hypothyroid, low calcium or phosphate diet, low iron, or is in chronic inflammation, they will probably have high 1,25 levels already from high conversion of 25. if u fix just 1 but still have the others it might not matter because they can all ramp up conversion
Then if someone adds more cholecalciferol , they are adding more 25, and they have more 25 to convert, they are adding fuel to the fire, and they are inhibiting mitochondria even more where it's already being inhibited by high 1,25. and if u add magnesium it increases 1,25 further because its needed for the converting enzymes (or if not enables it to work vs having lower amounts).
For me this explains the sometimes extreme effects well including twitching and heart effects from the lower t3 signalling, and also explains why magnesium made things worse
if someone is in hypothyroid state, taking something that either itself or at least what it converts to functions to further inhibit mitochondria, seems like a bad ideaan upside of it looks to be when it's in balance in a state of high respiration, preventing excessive respiration that would otherwise lead to dysfunction (paradoxically u could get lower mitochondrial function because of more damaged mitochondria otherwise, but this didnt effect the VDR knockout mice in this aspect it improved their mitochondria function for the timeframe tested), or used for enhancing proliferation / repair where needed by redirecting stuff from what would be used in the citric acid cycle
- vitamin D already converts to 1,25 highly in states of poor health , and 2. (if 25 vit D has some effects that gets around the VDR increase inhibiting thyroid receptors, not increasing VDR that much or something, or like ray mentioned in that audio, receptors act like quick accelerants for effects but arent the whole effect, 1,25 and 25 share a majority of gene expression changes i think >80% in common, but maybe there's enough differences idk) ,
-
This is a great thread for adding content to the idea that one can over supplement vitamin D. During the summer, I get my vitamin D the old-fahsioned way and my tan (aka solar calluous) makes it difficlut for me to get too much. Supplements can short circuit this feedback system.
-
It is exhausting connecting all the dots in this thread. Oftentimes, that is the way I start. The more I read the same thing, the morethe dots connect
, and eventually it starts to make sense.The key is to remember calcitriol to be like PTH, in that they are stress hormones, and that the lower they are, the better. Nothing wrong with them being zero. Everything good about them being zero as thst means there is enough calcium coming from food and that the calcium is mostly at the ecf and the ratio of extracellular calcium to intracellular calcium is where it should be at 15000:1 And this is because potassium is regulating very well calcium entry into the cell such that intracellular potassium :extracellular potassium is at a 35:1 ratio.
All brought about by very good acid base balance.
Brought about by optimal mitochondrial oxidation.
Helped by low infection and low toxicity in the body.
-
From an exchange with cool people:
Vitamin D (978-0-12-809965-0) - David Feldman
"It is now widely believed the enzyme [CYP27B1; calcidiol → calcitriol] exists in nonrenal tissues to boost local production of cellular 1,25(OH)2D3 in a paracrine/intracrine system. Such a role would suggest that cellular 1,25(OH)2D3 concentrations in extrarenal CYP27B1 tissues might be higher than in the tissues of the classical endocrine system, which depend on renally synthesized blood-derived 1,25(OH)2D3 at a concentration around 10^−10 to 10^−9 M (e.g., intestine, bone, parathyroid gland). In turn, the genes regulated in extrarenal tissues (e.g., macrophage, colon, prostate) might be a less-sensitive cell differentiation and antiproliferative subset, known to be regulated in cancer cell lines at 1,25(OH)2D3 concentrations of 10^−8 to 10^−7 M under cell culture conditions. A role for the extrarenal CYP27B1 is also consistent with the finding that serum 25(OH)D levels are associated with various health outcomes from bone health to cardiovascular health. In particular, low serum 25(OH)D levels are associated with increased mortality for colon, breast, and prostate cancer; increased autoimmune diseases and greater susceptibility to tuberculosis; increased cardiovascular diseases and hypertension. The presence of CYP27B1 in cells of the colon, breast, prostate, monocyte/macrophage, and vasculature could explain why serum 25(OH)D levels are so critical to the normal functioning of these tissues."
"Patients with hyperthyroidism (high serum levels of thyroxine, T4 and/or triiodothyronine hormone, T3) have been reported to have low circulating levels of 1,25(OH)2D [55,56]. Recent studies in mice with T3-induced hyperthyroidism showed markedly suppressed serum levels of 1,25(OH)2D and renal expression of Cyp27b1 [26]. Functional studies using PCT cells demonstrated the presence of a negative thyroid hormone response element (TRE) −50 to −20 bp upstream of the transcriptional start site of the CYP27B1 gene with this TRE overlapping the sterol regulatory element (1α-SRE) and TATA box [26]. Data in this study indicated that SRE-binding protein 1c acts as a transcriptional inducer of CYP27B1, but this effect is compromised following treatment with T3. Transcriptional repression effects of T3 on CYP27B1 appear to be due to thyroid hormone receptor (TR)α, and TRβ1 heterodimerizing with retinoid X receptor (RXR)α, to prevent binding of SRE-binding protein to its DNA target. In this way, T3 indirectly suppresses expression of CYP27B1 [26]."
"The serum phosphate concentration is another major regulator of renal 1,25(OH)2D production. In adult humans, dietary phosphorus restriction causes an increase in circulating concentrations of 1,25(OH)2D to 80% above control values, an increase not due to accelerated metabolic clearance of this hormone [38]. Dietary phosphorous supplementation has the opposite effect. Although the mechanism by which decreased serum phosphate increases renal 1,25(OH)2D production remains uncertain [39], it is clear that in humans the calcium-phosphorous-PTH axis cooperates to regulate the conversion of 25(OH)D to 1,25(OH)2D in the kidney. For example, decreased serum calcium concentrations are immediately registered by the parathyroid chief cell calcium-sensing receptors, which, in turn, relax inhibition of PTH production and secretion. The resulting rise in circulating PTH levels directly stimulates the renal 1α-OHase, while a PTH-mediated phosphaturic response and a subsequent decrement in the serum phosphate concentrations indirectly promotes 1,25(OH)2D production (Fig. 8.1)."
"In contrast to its renal counterpart, the macrophage 1α-OHase is unaffected by the stimulatory effects of PTH and phosphate [95,102]. The macrophage plasma membrane is not enriched with PTH receptors [103], and there is no evidence macrophages are responsive to PTH or PTH-related protein in terms of stimulating the protein kinase signaling pathways that are associated with induction of renal 1α-OHase. Similarly, the macrophage enzyme appears to be uninfluenced by changes in the extracellular phosphate concentration [95]. Moreover, exposure of activated macrophages expressing 1α-OHase to a calcium ionophore stimulates the hydroxylation reaction [104], while increasing the extracellular calcium concentration has the opposite inhibitory effect on the renal 1α-OHase [105]."
"Although effects of extracellular phosphate and serum FGF23 on macrophage 1α-OHase activity have yet to be documented, decreased expression of the enzyme has been shown in peripheral blood monocytes treated with FGF23 in vitro [106]. However, the general conclusion is that the key extracellular signaling systems for renal 1α-OHase activity are not heeded by the macrophage enzyme. This provides an explanation for why 1,25(OH)2D production by the macrophage in diseases such as sarcoidosis is not subject to the same negative feedback control that is mediated by a drop in the serum PTH concentration and an increase in the circulating calcium and phosphate level. By contrast, macrophage 1α-OHase activity is potently inhibited by antiinflammatory agents such as glucocorticoids which have little or no effect on the renal enzyme. In vivo this is likely to be due in part to the effects of glucocorticoids on macrophage differentiation and apoptosis. However, studies in vitro suggest that there is also direct inhibition of macrophage 1α-OHase activity by glucocorticoids [74]."
"Of the various bioactive cytokines concentrated in the alveolar space of patients with active sarcoidosis [77,78,130,131], interferon γ (IFNγ) was found to be the principal stimulator of the sarcoid macrophage 1α-hydroxylation reaction, with IFNα having minimal affect only at higher concentrations [74]. However, it is now clear that many other cytokines are also able to stimulate macrophage 1α-OHase. Recent studies have shown that interleukin-15 (IL-15) [132] and IL-32 [133] play pivotal roles in the induction of macrophage CYP27B1 during innate immune responses to bacterial infection. Elevated expression of IL-15 is frequently associated with inflammatory diseases, notably sarcoidosis [134]; as such, IL-15-mediated induction of 1α-OHase activity may provide a link between the regulation of 1α-OHase in normal innate immunity and the dysregulated 1,25(OH)2D production associated with granulomatous disease. Other cytokines such as tumor necrosis factor α (TNFα) [43,79] and IL-2 [102] are also know to stimulate macrophage 1α-OHase. As all of these factors have been implicated in the maturation of macrophage responsiveness within the innate immune system, it seems likely that upregulated 1α-OHase activity is a common feature of activated, but not quiescent, macrophages. More recently and in contrast to the effects of type 2 IFN, IFN-γ, it has been shown that type 1 IFN, IFN-β, negatively regulates the expression of 1α-OHase in human monocytes through increasing the expression of IL-10, which, in turn, inhibits CYP27B1 expression. This results in attenuated 1,25(OH)2D synthesis and consequent reduction in the antimicrobial peptide cathelicidin [135]. Similar observations have also been made for IL-4 that potently suppresses 25(OH)D-induced antibacterial responses in macrophages [136]. In this case, the precise effects of IL-4 on CYP27B1 are unclear, and may involve indirect effects via CYP24A1 [136]."
"Signaling via cytokines such as IFNγ may also lead to the activation of other calcium-dependent pathways in the macrophage, specifically the PKC [143] and phospholipase A2 (PLA2) pathways [144,145]. Because the macrophage 1α-OHase was not influenced by attempts to directly stimulate or inhibit PKC, attention has focused on the PLA2 pathway and the endogenous arachidonic acid metabolic cascade as the signal transduction pathway of most influence over the macrophage enzyme. Further dissection of the intracellular arachidonate metabolic pathway in this cell demonstrated that signal transduction through the 5-lipoxygenase pathway, specifically with the generation of leukotriene C4, was most critical to an increase in 1,25(OH)2D synthesis [146]. These studies were extended to investigate another compound with potential actions in the PLA2-arachidonic acid pathway, the 4-amino quinoline derivative chloroquine. Synthesis of 1,25(OH)2D by macrophages was completely inhibited by exposure to chloroquine in vitro [86]. Furthermore, this effect is independent of chloroquine’s apparent ability to alter the pH of intracellular organelles. When given orally to a hypercalcemic patient with sarcoidosis, chloroquine [83,86] or its analog hydroxychloroquine [84] can effectively reduce the serum 1,25(OH)2D and calcium concentration within 36 hours."
"Pathogen-associated molecular patterns such as the bacterial LPS are potent inducers of macrophage 1α-OHase expression and activity [95,102]."
"As outlined above, LPS and IFNγ commonly activate different signal transduction pathways but, as outlined previously, there is potential for cross talk between these pathways, which may have a significant impact on transactivation of CYP27B1. Notably, IFNγ and LPS are also the two most effective stimulators of nitric oxide (NO) synthesis in macrophages, and this has supported the hypothesis that production of NO and 1,25(OH)2D in macrophage-like cells may be functionally linked [151–153]."
"It is therefore interesting that two of the major stimulators of the human macrophage 1α-OHase, IFNγ, and LPS, are also key transcriptional regulators of the iNOS gene (NOS2) [156,157], which is itself a CYP-linked oxidase [158]. These observations coupled with the fact that NO has established inhibitory effects on other CYPs [159,160] suggest a possible link with the enzymes involved in vitamin D metabolism."
Cytochrome P450 Vitamin D Hydroxylases in Inflammation and Cancer
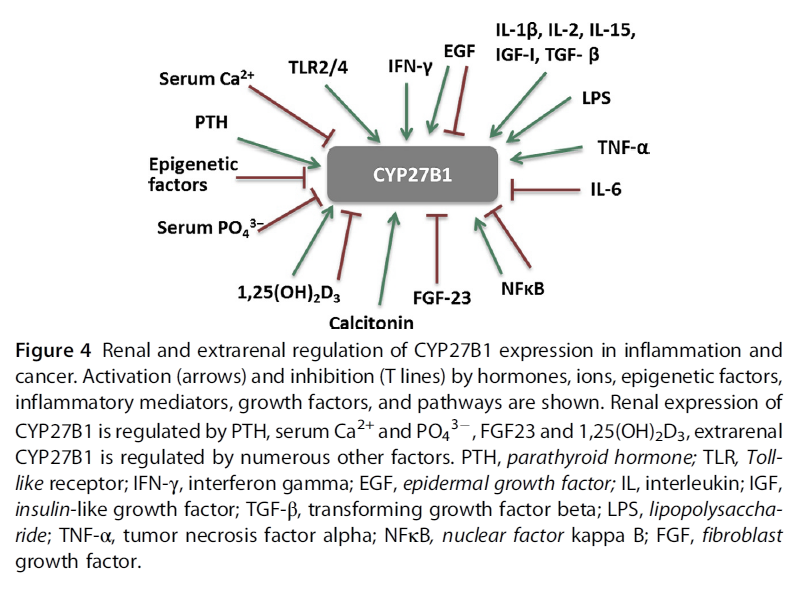
"Regulation of CYP27B1 expression is tissue dependent.
- In the kidney, the main regulators are PTH, serum calcium and phosphate, FGF-23, and 1,25(OH)2D3 (Armbrecht, Boltz, & Hodam, 2003; Perwad & Portale, 2011). Inhibition of CYP27B1 expression by 1,25(OH)2D3 involves complex epigenetic regulation of its promoter through VDR (Kim et al., 2009). However, there are tissues where 1,25(OH)2D3 either induces CYP27B1 expression or has no effect.
- In extrarenal tissues, CYP27B1 expression and activity are independent of circulating PTH. In prostate epithelial cells, the epidermal growth factor (EGF) induces, while 1,25(OH)2D3 inhibits CYP27B1 expression (Wang, Flanagan, et al., 2004). In macrophages and keratinocytes, the expression is increased by different cytokines, e.g., interferon gamma (IFN-γ), or tumor necrosis factor α (TNF-α) (Hewison et al., 2007), while nuclear factor kappa B (NFκB) is a potent inhibitor of CYP27B1 expression (Fig. 4)."
"There is evidence from in vitro studies that inflammatory cytokines influence expression of vitamin D metabolizing enzymes. While renal CYP27B1 expression is tightly controlled by PTH and 1,25(OH)2D3, in extra-renal tissues, CYP27B1 is regulated independently of those factors in a tissue-specific manner (Fig. 4). Soluble factors like cytokines and growth factors from the microenvironment affect cellular levels of this enzyme. IL1, IFN-γ, and TNF-α increase CYP27B1 expression in immune cells (van Etten, Stoffels, Gysemans, Mathieu, & Overbergh, 2008). TNF-α stimulated activity of CYP27B1 in untransformed cells such as human keratinocytes, endothelial cells (Zehnder et al., 2002), and peripheral blood monocytes (Gyetko, Hsu, Wilkinson, Patel, & Young, 1993). In cells of the human placenta, TNF-α induced expression of both CYP27B1 and CYP24A1, the latter to a greater extent (Noyola-Martínez et al., 2014). In the alveolar macrophages of patients with sarcoidosis, an inflammatory granulomatous disorder, CYP27B1 is expressed at a very high level, leading to pathologic increase of systemic 1,25(OH)2D3 levels and to hypercalcemia (Inui et al., 2001; Jones, 2008). In addition to the high CYP27B1 expression and activity, 1,25(OH)2D3-induced upregulation of CYP24A1 leads to the expression of a splice variant that lacks the mitochondrial targeting sequence thereby blunting the negative feedback regulation (Zehnder et al., 2002), contributing to the high circulating 1,25(OH)2D3 levels. CYP27B1 expression in macrophages is controlled by immune signals, such as IFN-γ, LPS, or by viral infections (van Etten et al., 2008)."
"In DCs, as in other extrarenal tissues, the level of the precursor 25(OH)D3 is often the limiting factor in the synthesis of the active metabolite (Jeffery et al., 2012). LPS, the ligand for TLRs, increased gene expression of CYP27B1 in DCs and human macrophages (Liu et al., 2006). 1,25(OH)2D3 synthesis in monocyte-derived DC is impaired due to a truncated, less active CYP27B1, while catabolism is not affected. Regulation of VDR targets in these cells seems to occur in a paracrine manner. It is the 1,25(OH)2D3 produced by macrophages that induces expression of vitamin D target genes in the neighboring DCs, inhibits maturation of DC, and reduces DC-dependent T-cell responses (Kundu, Chain, Coussens, Khoo, & Noursadeghi, 2014)."
"T-cell-derived cytokines regulate expression of the activating enzyme CYP27B1 in monocytes via TLR 2/1. IFN-γ increased the activity of CYP27B1 and decreased that of CYP24A1 (Noyola-Martínez et al., 2014). Mechanistic studies using promoter–reporter constructs in monocytes revealed that IFN-γ-induced increase of CYP27B1 is mediated via JAK– STAT, NFκB, and p38-MAPK pathways (Stoffels et al., 2006). Activation of TLR1 and 2 by Mycobacterium tuberculosis or IFN-γ increases expression of VDR and CYP27B1, (Adams et al., 2009; Liu et al., 2006). The resultant, locally synthesized 1,25(OH)2D3 induces the expression of antimicrobial peptides, such as cathelicidin (Gombart, 2009; Hoyer-Hansen, Nordbrandt, & Jaattela, 2010) or defensin 4β (Wang, Nestel, et al., 2004)."
-
@Amazoniac Thanks for the post. Ive been looking into those enzymes more and connected more things,
(*quick version: cyp 3a4 can get rid of excess 1,25, predisone can reverse extreme soft tissue calcification completely from this https://onlinelibrary.wiley.com/doi/full/10.1002/jbmr.51
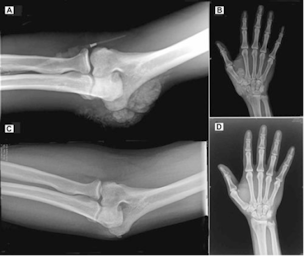
and chloroquine can reverse 1,25 excess too in 5 days, BUT big downsides to these.
if u are iron deficient with low cyp 3a4, upping iron can correct this and presumably normalise the excessive 1,25, which is probably why you see people on forums mention iron fixing their twitches along with the general improvement in ATP if fixing low levels)- among the main enzymes involved in 1,25 vit D degradation 2 are cyp24A1 & cyp3a4
BUT when it comes to macrophages CYP 24a1 is ineffective
- in response to stimulation with 1,25[OH]₂ D, upregulation of the inactivating enzyme CYP24A1 curtailed the functional effects of vitamin D in DCs, but not macrophages.
- rifampin fixes 1,25 excess in sarcoidosis through inducing CYP 3a4 enzyme
- So CYP 3a4 can be used to lower the 1,25 excess coming from macrophages
(another target would be lowering the increased CYP 27 A1 in macrophages) Macrophages showed a strong expression of CYP27A1, whereas monocytes and dendritic cells expressed low levels of CYP27A1 mRNA. Accordingly, macrophages converted vitamin D3 into the active metabolite 1,25(OH)2D3
side note 24,25 vit D increases bone strength / fracture repair where 1,25 does not have this effect https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6063485/
aside from impact on thyroid hormone Iron-sulfur clusters are a key part of the ETC needed to help the complexes I II III and part of the TCA cycle. i havent seen this pointed out much
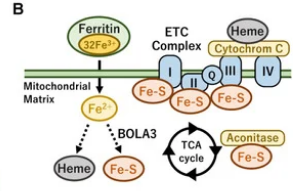
The iron–sulfur cluster is an essential component of the ETC complexes I, II, and III and aconitase in the TCA cycle. Therefore, mitochondrial iron metabolism is essential for mitochondrial respiration and the thermogenic capacity of brown adipocytes.
Within the electron transport chain, Fe–S clusters play a critical role in transporting electrons through Complexes I, II and III to cytochrome c, before subsequent transfer to molecular oxygen. Fe–S clusters are also among the binding sites of classical mitochondrial inhibitors, such as rotenone, and play an important role in the production of mitochondrial reactive oxygen species (ROS).
& we don't need full anemia for iron deficiency in tissues to have detrimental effect on mitochondria
thermogenesis goes down in mice / rats fed iron deficient diets even without anemia just mild drop in hematocrit and hemoglobin. with some insulin resistance seen too
https://pubmed.ncbi.nlm.nih.gov/34383942/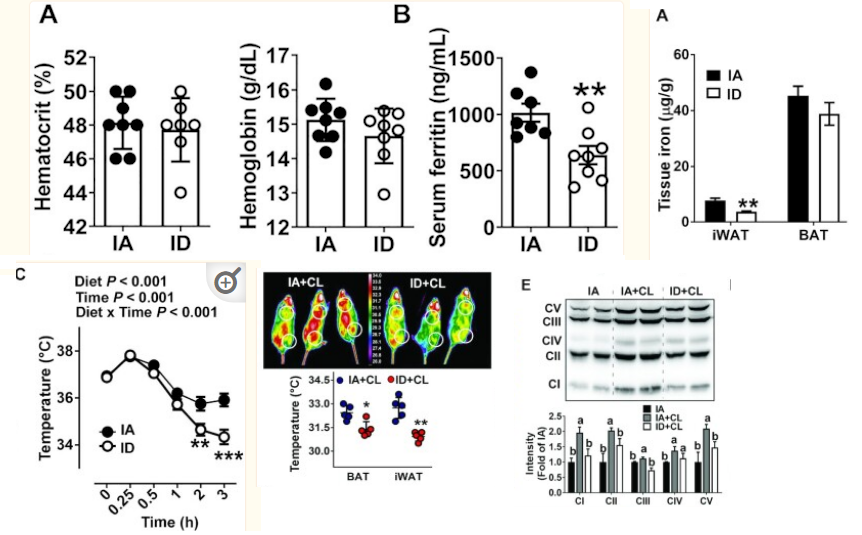
- Iron-deficient rats have increased blood and urinary catecholamines regardless of whether anemia is or is not present. The catecholamine response in both iron-deficient and control animals is largely temperature dependent, showing little difference at the isothermic {room} temperature of 30 degrees C but a two- to threefold increase in iron-deficient animals over controls at lower temperatures. The iron-deficient rat is unable to maintain body temperature at 4 degrees C and this is independent of anemia or of food intake. When animals are run on the treadmill for 4 h, body temperatures increase but the difference observed at 4 degrees C between iron-deficient and control animals persists. The underlying abnormality in temperature regulation and in catecholamine response disappeared after 6 days of iron therapy.
(also interesting: https://www.nature.com/articles/pr19852515.pdf Giving t3 to rats with iron deficiency (with anemia tho) the t3 restores grams of brown fat, but does not restore its activity, (ETC measures still the same, **** so taking t3 restores brown fat content but u still need enough dietary iron to enable t3 to exert effect on ETC ! )
The p450 enzymes (vit D metabolism) rely on a sulfur-iron protein adrenodoxin https://www.sciencedirect.com/science/article/pii/S0021925820410373
*** and here in humans https://pubmed.ncbi.nlm.nih.gov/18201586/
more than half the low iron people had low cyp3a4 , in these people restoring iron increased & near normalised their cyp3a4
= REGAINED ability to metabolise 1,25 if excessive(the only time ive felt restored in years was when i took iron consecutively, unable
A subgroup of 7 HD patients had significantly lower CYP3A4 activity before IV iron replacement compared with the other 5 HD patients and controls (mean [SEM] 0.86 [0.24] vs 2.30 [0.26] and 2.10 [0.26] (14)C exhaled/h; P < 0.01). After IV iron replacement, mean (SEM) CYP3A4 activity increased in these 7 HD patients (120.1% [67.1%]); P = 0.04) and it was not statistically different from that of controls (1.50 [0.36] vs 2.10 [0.26]).
-
- in response to stimulation with 1,25[OH]₂ D, upregulation of the inactivating enzyme CYP24A1 curtailed the functional effects of vitamin D in DCs, but not macrophages.
rifampin fixes 1,25 excess in sarcoidosis through inducing CYP 3a4 enzyme
- So CYP 3a4 can be used to lower the 1,25 excess coming from macrophages
side note 24,25 vit D increases bone strength / fracture repair where 1,25 does not have this effect https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6063485/
Insightful studies below that highlight 24,25 aka 24r,25 looks to be the commonly desirable form of vitamin D.
If in good health it's supposed to rise in ratio to 1,25 as 25 vit D goes up, https://www.nature.com/articles/s41598-019-43462-624r,25 form of vit D is likely protective in MS / brain atrophy https://pubmed.ncbi.nlm.nih.gov/21047880/ lower 24,25 compared to 25 = worse outcome
and it actually DECREASES the VD Receptor
Vitamin D3 is metabolized in the liver and kidney in addition to localized tissue-specific biosynthesis. Bone cells are known to metabolize 25(OH)D3 into both 1α,25(OH)2D3 and 24R,25(OH)2D3 via 1α-OHase and 24-OHase, respectively
24,25 DECREASES vdr expression and can lower 1,25,
it inhibits stem cell proliferation like 1,25 also does, but unlike 1,25 it stimulates stem cell maturation / enables their differentiation ,
increasing bone repair / strength https://pubmed.ncbi.nlm.nih.gov/24597546/(but by my experience cannot get this increased enough simply through vit D without worsening the issue from extra 1,25 in people with this excess issue,
was thinking maybe there isnt enough 24a1 enzyme producing 24,25 here, but upregulating doesnt work to counter 1,25 in macrophages)targeting less 1,25 through CYP 3a4 should have effect but also degrades 25 vit D though, so losing 24,25.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3310418/Thyroid hormone also increases cyp3a4
(another connection to hypothyroid & vitamin D problems)
and also decreases CYP27B1 , lowering 1,25 (at least in the kidneys, wonder if this also has effect in macrophages keratinocytes etc https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4077994/table/T1/?report=objectonly)lowering Cyp27b1 is a major target for lowering 1,25, it stays expressed even when macrophages are removed from IFN-y inducing environment https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4077994/
How was it that synthetic 1-hydroxylating activity in macrophages harvested from humans with active disease persisted ex vivo in culture even in the absence of conditioning IFNs? Some of the questions have been answered and some are still coming to light. For example, it is now known that there exists a single gene that encodes the 1-hydroxylase, CYP27B1 [11] and the 24-hydroxylase, CYP24A1 [12]. Further, it is now clear that persistence of mTB in macrophages is an explanation for the continual expression of the CYP27B1 in human macrophages even after their removal from the host and conditioning cytokines present in the inflammatory microenvironment in the host.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6508960/
(thyroid hormone & iron key players) & upping 24,25- In this work, we demonstrate that THs induce a significant increase in CYP3A4 mRNA levels, protein expression and metabolic activity through the membrane receptor integrin αvβ3 and the activation of signalling pathways through Stat1 and NF-κB.
-
This post is deleted! -
 L LucH referenced this topic on
L LucH referenced this topic on
-
@cs3000
You had mentioned relative lack of copper in this context of Mg and Zn and VDR activation making things worse and exacerbating underlying mitochondrial energy impairments.
Did you eventually supplement copper and did it profoundly fix these conditions for you?
Very interested in reading about this.
Thanks for having found and reported the 1,25D/VDR effect of inhibiting cellular copper uptake.
Have you made any approach in the meantime into resolving your underlying causes of elevated chronic inflammation/PAMPS/TLR?@cs3000 said in:
Th1 activity leads to paracrine extra-renal conversion of inactive 25D to active 1,25D, often depleting serum levels of measured 25D in order to increase active vitamin D (how can It deplete if theres 100x more 25 than 1,25 in plasma?) <←- NOT DIRECT DEPLETION , THE 1,25 EXCESS CAUSES ENZYME ELEVATION THAT METABOLISES 25
Afaik by drawing upon some of the same sources you've been using:
By measurements of serum levels (extracellular) of these hormones we only get a one-sided and distorted view on their balance between each other.
Since conversion to and activity of 1,25D are both predominantly intracellular processes there are concentration gradients to be accounted for.
I.e. only 1/10 of circulating serum 25D seeps across the cellular membranes and only 1/10 of intracellular 1,25D seeps outside, roughly. Both 25D3 and 1,25D3 are then essentially in the same potency range, could even be equimolar, with regard to exertion of their nuclear receptor binding intracellularly. -
i had similar symptoms than cs3000 described in the link above from drinking mostly raw milk and direct sun exposure(never had these symptoms from sunlight before i started drinking raw milk, and i have experienced these symptoms afterward in winter just from the raw milk), eating very good dattes temporary make me experience 0 bad effects from sunlight and if i have keep eating it at a certain point body even crave sunlight and sunlight exposure feel exhalting
-
Apparently, 25D which is the metabolite of D3, inhibits 1-25D production
“...With high dose cholecalciferol therapy... PTH is physiologically down-regulated and therefore the synthesis of 1,25-D and dietary calcium absorption are reduced.” — Vaidya, et al. (2015)" -
Methylene Blue should be able to inhibit 1,25 conversion too if hydroxychloriquine can.
-
@bio3nergetic Interdasting, we should get a list of inhbitors / factors that influence the conversion
-
Thanks @user1. Do you mean dates as in the fruit, like medjool dates? Because of their c. 0.3mg/100gr copper content?
I remember I had sort of kept myself afloat for many months by consuming a few hundred grams of dates every day and had assumed their delayed sugar release and balancing potassium content to be the major reason. -
@CrumblingCookie said in Vitamin D Receptor stops mitochondria respiration [Why vit D can cause problems] [1,25 vitamin D]:
Thanks @user1. Do you mean dates as in the fruit, like medjool dates? Because of their c. 0.3mg/100gr copper content?
I remember I had sort of kept myself afloat for many months by consuming a few hundred grams of dates every day and had assumed their delayed sugar release and balancing potassium content to be the major reason.Yes this fruit, yet much softer and moistyer thab typical medjool or dattes
-
@alfredoolivas Progesterone lowers 1,25 as well
-
Is it possible to have copper deficiency, but normal levels of serum copper and ceruloplasmin, and serum free copper abnormally high?
-
@BearWithMe estrogen can increase free copper while PUFA can increase estrogenic activity in inducing free copper or directly damaging enzymes related to copper utilization. Free copper is what contributes to toxicity. Total copper in a healthy system is what yields robust copper status.
Hello! It looks like you're interested in this conversation, but you don't have an account yet.
Getting fed up of having to scroll through the same posts each visit? When you register for an account, you'll always come back to exactly where you were before, and choose to be notified of new replies (either via email, or push notification). You'll also be able to save bookmarks and upvote posts to show your appreciation to other community members.
With your input, this post could be even better 💗
Register Login