All things Histone Deacetylase (HDAC) and DNA Methyl Transferase (DNMT) inhibitory - reversing epigenetics from metabolic insults
-
Although I wouldn't decline some vorinostat aka SAHA aka suberoylanilide hydroxamic acid or romidepsin I'll go for the non-pharmaceuticals first:
• I am about to get the whole range of available butyrates; Na-But, Ca-But, Mg-But, K-BHB! And hopefully those will be as effective and pervasive into tissues as phenylbutyrate (without the nitrogen/glutamine/BCAA-depletion of the latter) or even better at an equivalent daily dosing between 10-20grs. The BHBs are and add-on for bOHbutyrylation, of the histone site H3K9, predominantly.
The different HDAC enzymes all preferably target different sites of the lysine chains.
• Choosing between sulforaphan (SFN) or erucin or isothiocyanates I'm going for EGCG to inhibit DNMT activity because the latter, in comparison, has the extra benefit of shuttling zinc across membranes and binding free iron instead of "merely" upregulating ferritin and intracellular metallothionine deposits. That action against free iron should prevent ferroptosis in healthy cells and increase intracellular TET proteins (which oxidize DNA methylation).
• Furthermore, maintain GSH (against hypermethylation) with extra NAC and
• maintain sufficient zinc, but not in excess!? Yet enough to competitively displace an overly abundant ratio of free iron and copper?*.
• As well as sufficient B2 and folate which are necessary for binding to a FAD‐dependent histone demethylase named lysine‐specific demethylase‐1 (LSD1). Folate is a necessary scavenger of formaldehyde which is being created by demethylization and therefore a coenzyme for demethylization enzymes as DMGDH, SDH in the liver.
• I'll avoid niacinamide because of its inhibition of PGC-1α through inhibition of SIRT1 (and also SIRT2) and rather use niacinic acid? And keep B5 at a sane, low amount to not overexpress CoA which is said to speed up the reaction rates of HDAC1 and HDAC2?Would that be a solid approach?
Other HDACi:
• Btw butein looks good, too, but it also being a very potent tyrosine kinase inhibitor (decreases mitogenesis, cell division and cell proteins) sort of predestinates it for use in cancers with/by overactive TK activity rather than for non-anticancerous use, I guess. Class I, II, IV HDACi.• Puerarin looks interesting as it stimulates AMPK, PGC1α, Nrf2.
Increases autophagy, reduces Apoptose (PI3K/Akt activation = GSK3beta inhibition).
Inhibits mTOR, TNFα, NF-κB, JNK, Cox2, HDAC1 and HDAC3
Increases PPARy though which is nothing critical but not nice long-term.• Apigenin or the congeneric Luteolin ?! Inhibition of DNMT and HDAC class I and, more weakly class II.
• Decursin, which is a coumarin-derivative from Angelica plants. Predominantly DNMTi (looks for for synergy with a HDACi).
Orphan drugs or still in pharmaceutical trials:
• Panobinostat (cancelled in US, available in EU for >€4100/6 tablets): class I, II, IV HDACi
• (Mocetinostat aka MGCD0103, inhibits mainly HDAC1, plus 2/3/11)
• (Entinostat aka MS-275 (patented), inhibits class I HDAC1 and HDAC3 with IC50 of 0.51 μM and 1.7 μM, respectively.)
• (Belinostat aka PXD101, only i.v.): class I, II, IV HDACi
• Vorinostat aka SAHA aka suberoylanilide hydroxamic acid: class I & II HDACi
• Romidepsin, HDAC1,2 + HDAC4, 6 (weaker): class I, (II) HDACi
• Valproic acid: class I HDACi. Very weak against class II.• Biotin: HDACi. Yet IME in a very specialized way because it's exacerbating when not in combination with a broad-range base.
• Vitamin E (which forms exactly?): HDACi. Yet IME in a very specialized way because it's exacerbating when not in combination with a broad-range base.• Possibly alkanes like:
Terpenes such as Phellandren, α-Terpinen, Limonen, Undecan- Resveratrol increases SIRT1. It's reported to inhibit DNMT1 and DNMT3b. It's a dirty, double-edged substance, imo, with a wide range of opposing effects. I don't like it much. It's kind of like eating super tasty icecream that has laxatives mixed into it.
Background info bits:
• The traditional DNA hypomethylating agents (HMAs) such as decitabine (DAC) and azacytidine (AZA) are super toxic and destructive.
• "The DNA methyltransferase (DNMT) family is comprised of the catalytically active DNMT1, DNMT3A, and DNMT3B, which methylate cytosines within CpG dinucleotides, as well as DNMT3L, which lacks catalytic activity and serves as a regulatory factor. Using S-adenosyl-l-methionine (SAM) as the methyl donor, DNMT3A and −3B establish the DNA methylation pattern de novo while DNMT1 is primarily responsible for propagation of this methylation pattern to daughter strands following DNA replication. Thus, DNMT1 preferentially binds to and methylates DNA containing a hemi-methylated CpG dinucleotide."
• On DNMT and HDAC interrelations: !
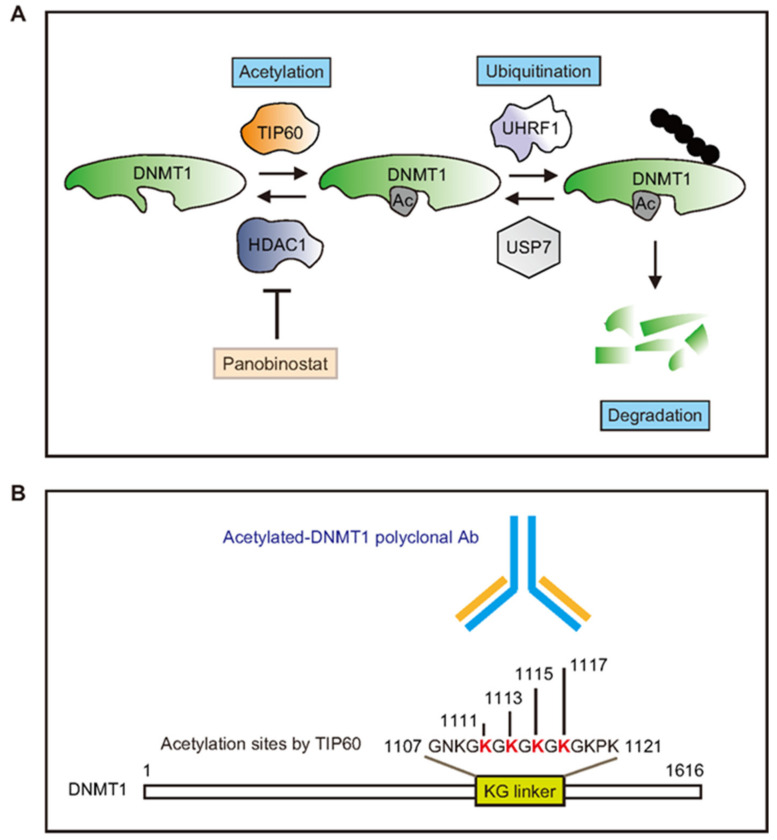
-> DNMT1 is acetylated by TIP60 promoting its degradation. TIP60 regulates DNMT1 acetylation, while histone deacetylase 1 (HDAC1) controls DNMT1 deacetylation.• GDNF, BDNF etc are all influenced by inhibiting HDAC.
• Here's a bit on the decisive short-term benefits of proper vitamin A; Food for thought for everyone who feels acute benefits from Retinyls:
"retinol/retinoic acid (vitamin A) and ascorbate (vitamin C) synergistically diminish DNA methylation levels and in doing so enhance the generation of naïve pluripotent stem cells.
This is achieved by two complementary mechanisms. Retinol increases cellular levels of TET proteins (which oxidize DNA methylation), whereas ascorbate affords them greater activity by reducing cellular Fe3+ to Fe2+" [10.1073/pnas.1608679113, 2016]Some papers:
• Targeting Histone Deacetylases with Natural and Synthetic Agents: An Emerging Anticancer Strategy, 2018
• Histone Deacetylases Inhibitors in Neurodegenerative Diseases, Neuroprotection and Neuronal Differentiation, 2020
• *: Therein, wrt zinc the HDAC familiy classification:
• And from here the same with descriptions of the respective HDAC enzymes' main targets:
• Metabolism, HDACs, and HDAC Inhibitors: A Systems Biology Perspective, 2021
• Function and mechanism of histone beta-hydroxybutyrylation in health and disease, 2022
@CrumblingCookie said in Random, interesting studies:
@cs3000 said:
something interesting,
when thyroid receptors aren't bound by the hormone / agonist they block DNA transcription. using HDAC. HDAC inhibition is 1 way to help the people who have receptor mutation / resistance, (sort of)
https://academic.oup.com/hmg/article/23/10/2651/614693#10263757
https://scholars.mssm.edu/en/publications/histone-deacetylase-inhibition-reduces-hypothyroidism-induced-neu
So basically people can get some of the gene effects from T3 activation if its lacking, without the t3 , by hdac inhibition . not full effects but someBam! So the working mechanism of T3 is not directly cellular stimulation, but inhibition of the inhibition of cell metabolism?
Therefore, vice versa, a practically hypothyroid state would be mimicked by overmethylation of the CpG sites on the DNA (blocking the transcription of genes) or by deacetylated/dephosphorylated/demethylated/de-beta-hydroxybutyrylated sites of the lysine chains of the histones (compressing the histones which wrap the DNA strands which prevents DNA access)?To supplement thyroid hormones would work in such circumstances but be a kind of force-feeding, rawhiding override and circumvention of the actual underlying culprits?
But using other inhibitors of HDAC or DNMT could then be actually better and closer to the original cause and also effectively act like thyroid hormones?I used to think of HDACis only as some very beneficial class of substances in a vague context of cancer (even though even in that they are very restricted).
Now they appear much more crucial in all kinds of diseases and chronic impairments.
If I were casually being offered some pure quality HDAC inhibitors I would gladly take them and run a treatment course with them.
@alfredoolivas said in Random, interesting studies:
CrumblingCookie HDAC inhibition also increases expression of thyroid receptor itself.
@Mauritio said in Random, interesting studies:
@CrumblingCookie Good point .
Well, sodium butyrate is widely available ... -
@CrumblingCookie

https://raypeat2.com/articles/articles/protective-co2-aging.shtml,
For the butyrate forms i think its better to avoid coated forms for general use, the ones that have fats coating them (small intestine is probably vulnerable to increased amounts released there https://pmc.ncbi.nlm.nih.gov/articles/PMC11641654/#sec2-ijms-25-12998, plus the undiluted sodium when coated, normal form gets absorbed higher)
and only small amounts needed < 200mg as sodium butyrate for h3 acetylation
doi.org/10.1111/bph.13637
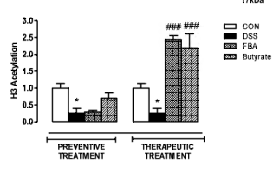
-
Thanks for this. I've just read the stressed mice study about first decreased, then significantly increased BA levels in the ileum, alongside drastically decreased free amino acids and a microbiotic shift.
So actually increased (ileal) BA levels associated with bad outcomes... ileal bacterial dysbiosis and overgrowth... Free-amino-acid starvation of beneficial ileal bacterial species... Due to lack of digestive functions in the more proximal parts of the GI system? Which more BA in the proximal parts would prevent and heal? Hence indeed the preference for uncoated BA instead of slow-release formulas as you suggest. I'll heed that founded advice.
At first, that study turned me somewhat angry for contradicting the model about lack of BA. But I take it's a cascade of which it is best to start at the top.Most current studies suggest that the intestinal mucosal barrier damage caused by CUMS is associated with low BA levels in the intestinal lumen. However, some reports described high fecal BA concentrations in CUMS model mice [21]. Some patients with mental disorders such as depression and autism also exhibit high intestinal BA concentrations.That study actually touches on and summarizes so many crucial elements of health. Not least when thinking about sprue or disaccharide intolerances or the vaguely ominous biogenic amines burden. Look at those dwindling goblet cells and exploding serum LPS and villous atrophy:
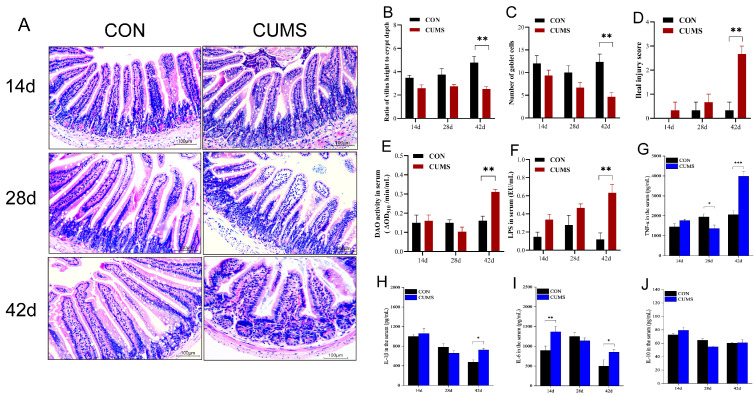
In the black-and-white bar graph above: Obliterated H3-acetylation by chemical injury (DSS).
Where did you derive or read the <200mg Na-But dose from? In that mice study they used 20mg∙kg−1.The importance of butyrate supplementation has been demonstrated by the impaired butyrate metabolism in the inflamed, intestinal mucosa of patients affected by IBD (De Preter et al., 2012). In fact, data show that this deficiency in butyrate results from a reduction in butyrate uptake by the inflamed mucosa due to the down-regulation of the monocarboxylate transporter (MCT)-1 expressed on the apical membrane of intestinal epithelium (Thibault et al., 2007).Reduced BA uptake in the chronically inflamed states. So it'd be better to really blast up the dosage? Perhaps even so much as to provide BA through serum circulation and the basal membrane (from within the body) instead of the apical side (from the intestinal lumen)?
From Protective CO2 and aging:
Prolonged lack of CO2, i.e. also hypoxic states -> epigenetic silencing/hibernation. Got it! That seems sensible to apply!I'm not fan of methionine. E.g. a high methionine, low cysteine ratio in many vegan protein powders feels very unsatisfactory for me. Betaine or trimethylglycine don't feel well either. Accordingly, red beets are a horror. IME, after a 2-3 days' subjective boost, those methyl donors can really worsen things. Yet merely avoiding their overabundance is insufficient in itself to put things back on a right track. The same with the idea of nicotinamide to deplete methyl-groups: That never worked for me. Choline however is really important for liver functions, chylomicrons etc. That thinking about the absolute dietary supply of methyl groups must either be way too simplistic or a mere drop in the ocean of stronger factors.
Speaking methylation: That's an ambiguous mechanism because whilst we don't want excessive methylation of DNA CpG sites which prevent genetic transcription, methylation of the lysine groups on histone bodies either contributes to their unfolding OR to genetic silencing.
On H3K9 tri-methylation (reportedly a stubbornly long-term, transgenerational impact!) acts silencing so directly opposes the effects of acetylation or bOHB. Which makes HDACis like butyrate or beta-hydroxybutyrate direct antagonists to overmethylation. I wish Peat had followed that up.
It could be rewarding to find out about the histone demethylation mechanisms for H3K9 specifically. -
@CrumblingCookie said in All things Histone Deacetylase (HDAC) and DNA Methyl Transferase (DNMT) inhibitory - reversing epigenetics from metabolic insults:
free-amino-acid starvation of beneficial ileal bacterial species... Due to lack of digestive functions in the more proximal parts of the GI system? Which more BA in the proximal parts would prevent and heal?
However, some reports described
high fecal BA concentrations in CUMS model mice [21]. Some patients with mental disorders
such as depression and autism also exhibit high intestinal BA concentrations.Where did you derive or read the <200mg Na-But dose from? In that mice study they used 20mg∙kg−1.https://pubmed.ncbi.nlm.nih.gov/25240644/ here valproic acid induced autism-like defects when given during pregnancy , but weirdly this or sodium butyrate fixed them when given for 5 weeks a while after birth. (affects folate during pregnancy idk if that applies to butyrate too, but maybe best to avoid HDAC inhibitors during)
np interesting thread & the 20mg/kg dose for HDAC inhibition orally for mice is about 100mg - 150mg for human when u account for metabolism differences. its not exact depends on substance but generally for mice its (3/37) * dose * human weight in kg , or just dose *5 to simplify for 60kg person and add or reduce as needed for weight. for rats 6/37 or dose *10
useful link for different animals conversion https://www.westernu.edu/media/research/iacuc/dose_conversion_between_animals_and_human_-jbcp_2016.pdfFrom Protective CO2 and aging:
Prolonged lack of CO2, i.e. also hypoxic states -> epigenetic silencing/hibernation. Got it! That seems sensible to apply!
Speaking methylation: That's an ambiguous mechanism because whilst we don't want excessive methylation of DNA CpG sites which prevent genetic transcription, methylation of the lysine groups on histone bodies either contributes to their unfolding OR to genetic silencing.
On H3K9 tri-methylation (reportedly a stubbornly long-term, transgenerational impact!) acts silencing so directly opposes the effects of acetylation or bOHB. Which makes HDACis like butyrate or beta-hydroxybutyrate direct antagonists to overmethylation. I wish Peat had followed that up. It could be rewarding to find out about the histone demethylation mechanisms for H3K9 specifically.ah key context there then , here global methylation was actually lower in hibernation https://pubmed.ncbi.nlm.nih.gov/25908059/ with a global transcriptional decrease https://pubmed.ncbi.nlm.nih.gov/19412897/ with increase in HDAC too especially hdac1 and 4.
(hibernation i think reflects as a darkened hellish depression state for humans, humans dont hibernate so the closest thing is creating a drive towards death. sleep isnt hibernation hibernation is closer to death just on the edge)
Reduced BA uptake in the chronically inflamed states. So it'd be better to really blast up the dosage? Perhaps even so much as to provide BA through serum circulation and the basal membrane (from within the body) instead of the apical side (from the intestinal lumen)?
thats what im thinking too the underneath part, gonna try standard butyric acid diluted in water, into circulation and see if helps a small intestine problem at the ileum from the underside. but some will absorb direct too. i think taking it uncoated = you still get it absorbed a lot in small intestine but spread out absorption in duodenum jejenum ileum and diluted with water, instead of overwhelming just the ileum and with the undiluted sodium release too as extra that doesnt release until then otherwise
1g+ looks like high dose used for acute repair boost and general use lower, (for histone acetylation they showed its effective systemically in tissues at low dose <200mg as the sodium, because most of the sodium butyrate wouldnt havent reached that colon tissue sample direct, so if bumping up 50% to make room for absorption issues still <300mg or 2x <500mg generally) -
It could be rewarding to find out about the histone demethylation mechanisms for H3K9 specifically.
It looks demethylation of H3K9 is a job of the LSD=KDM class of histone demethylases.
So the aforementioned folate- and FAD-dependent LSD1 is =KDM1A and demethylates H3K4me2/me1.
H3K9me3 demethylination is trough KDM4A, specifically:

This table's right column reveals whether demethylation activates or represses genes.
from Vitamin D and the epigenome, 2014
This paper was a chance find for me when looking for H3K9me3.
-> There are some serious implications on HDAC and epigenetics through ligand-activated VDR by 1,25-OH-D3 (or its ligand-inactivation by 25-OH-D3). The details of it depend a lot on co-conditions. In general, activated-VDR does not directly boost overall HDAC levels, but
it strongly recruits HDAC enzymes to specific histone sites. And it prevents KDM histone demethylation.
It offers necessary DNMT1/3B inhibition on promoter genes of innate immunity.
That HDAC recruitment is really crappy.
E.g. HDAC1 strongly prevents cell differentiation.
It fits in with e.g. a high density of VDR in irritable bowel disease and the general observations of forcing a higher 25-OH-D3/1,25-OH-D3 ratio by exogenous D3-supplementation (also topically) to bring about alleviating, healing, pro-metabolic effects on epithelial cell lines of e.g. skin, GI, airway surfaces.(Preexisting) HDAC3 induction strongly suppresses 1,25-OH-D3 synthesis and VDR expression and therefore innate immunity.
1,25-D3 inhibited the expression of several histone demethylases (e.g., KDM4A/4C/4D/5A/2B, JMJD5/6, PLA2G4B), and induced the expression of others, JARID2 and KDM5B (Pereira et al., 2012). Members of the KDM4 family catalyze tri-demethylation of H3K9 and/or H3K36 (Cloos et al., 2006; Fodor et al., 2006; Klose et al., 2006; Whetstine et al., 2006; Lin et al., 2008). H3K9me3 is a mark for heterochromatin and demethylation of H3K9 is suggested to be linked with chromosomal instability (Cloos et al., 2006). Inhibition of expression of KDM4 family members by 1,25-D3 could thus contribute to genome stability. The histone deacetylase HDAC3, one of the most frequently upregulated genes in cancer, seems to inhibit VDR expression. [...] As early as 1984, Yoneda et al. (1984) have shown that the histone acetyltransferase inhibitor butyrate augments 1,25-D3 actions. Several studies confirmed these findings (e.g., Rashid et al., 2001) and have suggested that the action of butyrate could be through upregulation of VDR or CYP27B1 expression (Gaschott and Stein, 2003). Whether this effect is mediated by direct acetylation of the VDR or CYP27B1 promoters, has not been determined.Here's another paper:
Vitamin D opposes multilineage cell differentiation induced by Notch inhibition and BMP4 pathway activation in human colon organoids, 2024
-> No explicit mentioning of HDAC or DNMT, but pointing towards change of epigenetics expression
-> Limited cell renewal with high VDR-activation -> more stem-cell reserves.
But what's the use of withholding stem cells? Statically, I can't think of a benefit. Situationally, it could be very beneficial i.e. as in maintaining low 25-OH-D3 = high VDR-activation before chemotherapies, then stimulate stem cell differentation to quickly replace damaged tissue after chemotherapy.In wonder what other substances stimulate (specific) KDM histone demethylases.
@cs3000 said in All things Histone Deacetylase (HDAC) and DNA Methyl Transferase (DNMT) inhibitory - reversing epigenetics from metabolic insults:
(hibernation i think reflects as a darkened hellish depression state for humans, humans dont hibernate so the closest thing is creating a drive towards death. sleep isnt hibernation hibernation is closer to death just on the edge)
That's an oddly-terrifying novel description for a subjective state of being. A metabolic stasis. Hibernation torpor, metabolic depression, apnoic breathing, hypoxia, blurred states of consciousness and bodily functioning all don't sound too well indeed.
Good finds: In this first paper you linked they observed overall CpG-hypomethylation in hibernation. As for histones, they referred to a prior 2006/2009 paper of which you quoted the summary of table 1: Increased HDAC activity (x1.8), decreased H3S10 phosphorylation (x0.61), decreased H3K23 acetylation (x0.75). And, unsurprisingly, much reduced genetic transcription.
What do we take from this? That DNMT inhibition is crucial for innate immunity and anti-cancer genetic expression but the dismal hibernation is mediated mainly through increased HDAC? So HDAC activity really sets the metabolic quality of life.
And maybe, in some circumstances, low DNA methylation (beneficial) could even go hand in hand with high histone/chromatin compaction (dismal)?
Unless there's a smart combination or at least compensation of pathologically deranged factors.Smart considerations could be to regard year-round continous D3 supplementation as a misuse for it's HDACi + KDM side effect and it to be better to rather choose a HDACi. Or VDR stimulation + butyrate HDACi.
-
In contrast to kvirion's 1g daily dosing idea,
I had started with 2grs of Na-butyrate, 4-5 times a day (i.e. 8-10grs per day in total) on Wednesday. Since Thursday my skin and body has been feeling warmer and better perfused.
Most noticably, on Thursday and Friday, all off a sudden I had functioning GI mucous layers again: Whilst bowel movements were frequent, the stools were the smoothest I've had in years if not ever. So smooth that no Tp was necessary at all.
Then Saturday and Sunday that strongly reverted to the very opposite. I could even smell some butyrate as a cause or a consequence of this disturbance.I've then decreased the dosage to 1g NaB four times a day.
Now my thoughts are:
Did I overdo the butyrate dosages and by this deplete necessary cofactors,
or did I overdo the butyrate and do its beneficial effects maybe follow a Gaussian distribution curve. I.e. is there an ideal, optimum dosage range for butyrate, with higher amounts reverting much of its benefit?
In other words, is there an inverse dose response past a certain amount?
In a human study with 4-phenylbutyrate 9g/day increased to 21g/day over 24 weeks, Phase 2 study of sodium phenylbutyrate in ALS, 2009,
"The greatest increase in histone acetylation occurred at the lowest dose (9g/day) but did not reach the levels of normal individuals – the authors were unclear why an increase in histone acetylation was highest at the lowest dose".
There were 40 participants but only 26 of them completed the trial through the dosage increments. Because the daily dosage was increased weekly it's also not possible to distinguish the effects between daily dosage and prolonged drug exposure or disease progression.
participants took three tablets t.i.d. from the baseline visit to week 2 (9 g/day), four tablets t.i.d. from week 2 to week 4 (12 g/day), five tablets t.i.d. from week 4 to week 8 (15 g/day), and six tablets t.i.d. week 8 through week 12 (18 g/day). At 12 weeks, the subject took seven tablets t.i.d. (21 g/day) through week 20, the last day of the treatment phase of the trial (Figure 1). If participants could not escalate dosage because of adverse events, they were maintained on their maximum tolerated dosage.In figure 3 the authors show Histone acetylation levels with NaPB dose taken prior to the blood draw. The peak is clearly highest (but perhaps even post-maximum?) at the lowest trialled dosage of 3g NaPB (=1.4g butyrate = 1.8g Na-B) and significantly declines at higher doses of 4g or 5g, respectively.
4-phenylbutyrate contains 53% butyrate by weight.
Sodium 4-phenylbutyrate (used in the ALS trial above) contains 47% butyrate.
Sodium butyrate contains 79% butyrate by weight.
Calcium butyrate contains 81% butyrate by weight.
Magnesium butyrate contains 88% butyrate by weight.The 9g Na-PBA used in the ALS trial therefore equals 4.23g butyrate, as in 5.35g Na-butyrate per day.
If aiming at such a maximum amount of butyrate per day, this accordingly equals
• 5.2g calcium butyrate which also provides a practically convenient amount of 992mg calcium,
• 4.8g magnesium butyrate which also provides a very substantial 577mg magnesium.A 5:1 or 4:2 blend of Ca-B to Mg-B could be good over the long term:
Max. 5g of a 5:1 blend yields c. 790mg calcium, 100mg magnesium, 4.1g butyrate.
Max. 5g of a 4:2 blend yields c. 630mg calcium, 200mg magnesium, 4.2g butyrate.
I like how this can beneficially cover extra Ca and Mg needs in contrast to the equivalent extra sodium, which is probably an unneccessary burden to most people except for those with very speficic kidney issues. -
This post is deleted! -
@herenow said:
You could end up with an effect similar to Rx medications by combining weak inhibitors and the drugs seem to have toxicity. It could be an issue over time.
Do you mean that the the non-Rx substances would, especially in combination, clearly be safer and similarly effective than taking Rx medications?
Or do you mean that the non-Rx substances could show similar toxicities like Rx over the course of time?There seems to be a sweet dosage range for best effects. Overdoing the inhibitors could deplete stem cells through ↑cell differentiation , ↓ proliferation and ↑ apoptosis.
As in the case with those butyrate-sensitive ISCs which are meant to be particularly protected at the bottom of special intestinal crypts.@herenow said:
The only established application for the drugs that I could find is cancer treatment and I did not yet find many off label uses.
Yes the medical guidelines and practices are very restrictive about them. Even if you look up the cancer applications you'll see those Rx inhibitors are only being allowed to patients after a failed first-line round of chemotherapy and then only in combination with another bout of chemotherapy.
But the hints and references to epigenetically engrained alterations are everywhere.
Not least in psychology and psychiatry where it's known that e.g. in depression epigenetic expression and methylation patterns are all over the place in significant deviation from "healthy" controls.In the vitamin D study linked above e.g. they wrote of dibenzazepine as stimulating specific lines of cell differantion [to goblet cells in the case of the GI system], which is interesting because that whole dibenzazepine drug class includes (ox)carbamazepine and a hole range of tryclic neuroleptic and antidepressants and constitutes one of the cornerstone of hit-and-miss psychiatric medications with a very wide range of effects and oven irresponsibly prescribed to "reset" incomprehensible brain metabolism.
And many probably know of the countless rodent experiments in which animals were stressed in various ways and various developmental stages and then went on to produce offspring exhibiting transgenerationally lasting disturbed behaviour, depression and giving up early in those notorious forced swimming tests across 5, 6, 7 or more generations.
You are right about recognizing this is a still very blank spot on the map of the therapeutics world. I suggest there is treasure to be found.
-
This post is deleted! -
This post is deleted! -
This post is deleted! -
baicalin can ameliorate neuropathic pain by suppressing HDAC1 expression https://pubmed.ncbi.nlm.nih.gov/23684218/
-
@Mauritio said:
baicalin can ameliorate neuropathic pain by suppressing HDAC1 expression https://pubmed.ncbi.nlm.nih.gov/23684218/
Yet it enhances HDAC2 and inhibits histone demethylase (LSD1) at an IC50 of 3.01μM.
The LSD1 inhibition can be either a welcome or unwelcome dual-function.
https://pubmed.ncbi.nlm.nih.gov/22421405/
https://pubmed.ncbi.nlm.nih.gov/27814566/
I wouldn't know when to use baicalin and when not beside the neuropathic pain scenario.
There has actually been research into a series of dual HDAC and LSD1 inhibitors via conjugating the LSD1 inhibitor tranylcypromine to various o-aminoanilide and hydroxamic acid-based HDAC inhibitors, preferably with vorinostat by Duan et al., 2017 and with entinostat by Kalin et al., 2018.
Domatinostat is by itself already an class I HDACi and LDS1i. Those bifunctional HDACi+LDS1i compounds appear to be significantly more effective against cancer.Specifically against cancer baicalin works through a different pathway: Inhibition of hexokinase II which is located at the outer mitochondrial membranes and provides the cancer cell's excessive glucose needs.
-
Apigenin and the congeneric luteolin are potent class I HDACi, especially strong against HDAC2 and HDAC8.
Much less affinity to HDAC7, which would be targeted by class IIa HDACi (HDAC4/5/7/9).
[The more negative the value of docking score, the higher is the affinity of ligand toward receptor.]On a simple by-weight calculation this could convert to 100-150mg apigenin/day for a human:
"Moreover, oral intake of defined flavone (20 and 50 μg/mouse/day) for more than 8-week period has revealed a substantial reduction in tumor growth of PC-3 xenografts in athymic nude mice [41]."
From: In silico approaches for investigating the binding propensity of apigenin and luteolin against class I HDAC isoforms
Luteolin, promising a very good safety profile, may alleviate hyperalgesia, chronic inflammatory or neuropathic pain. The authors make no mention of HDAC or epigenetics at all but point to luteolin's strong anti-inflammatory and antioxidant properties. Ultimately it may well be due to inhibitions of HDAC, similar to and in line with the aforementioned baicalin:
Luteolin: A promising natural agent in management of pain in chronic conditions
-
This in-silico modelling shows butyric acid being also an inhibitor of class IIa (HDAC7). It also reveals that phenylbutyrate does indeed exert stronger action:
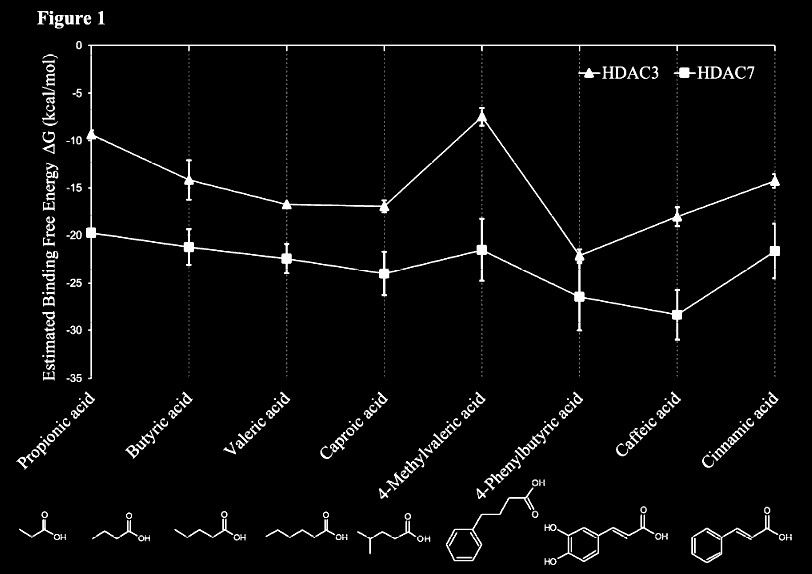
From: In Silico and in Vitro Interactions between Short Chain Fatty Acids and Human Histone Deacetylases
-
I've seen apicidin [cyclo(N-O-methyl-l-tryptophanyl-l-isoleucinyl-d-pipecolinyl-l-2-amino-8-oxodecanoyl)] in a graphic put into the same HDACi group of cyclic peptides along with romidepsin and briefly followed up on that one:
It appears apicidin is orally and parenterally effective in vivo against protozae like plasmodium (malaria) by inducing hyperacetylation of histones in treated parasites. Potentially also active against toxoplasma, cryptosporidia etc.
"Other known HDA inhibitors were also evaluated and found to possess antiparasitic activity, suggesting that HDA is an attractive target for the development of novel antiparasitic agents."
Apicidin: A novel antiprotozoal agent that inhibits parasite histone deacetylase
"Treatment of P. falciparum cells with 70nM of apicidin at these three developmental stages resulted in ∼90% reduction of growth (IC90)"
Histone Deacetylases Play a Major Role in the Transcriptional Regulation of the Plasmodium falciparum Life Cycle
-
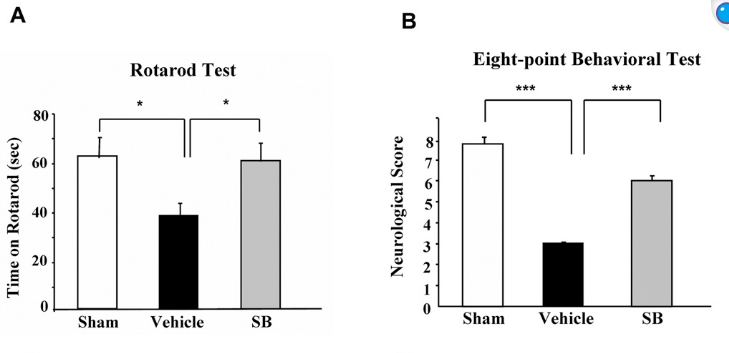
Looked into the effect of sodium butyrate on causing senescence / stopping cells proliferate. it can do that to normal fibroblasts (even tho showed increased wound healing). but effect is selective
pretty good one showing this helps recovery after brain injury , https://pmc.ncbi.nlm.nih.gov/articles/PMC2726719/#S10
during continued poor blood flow to the brain it helped restore brain cells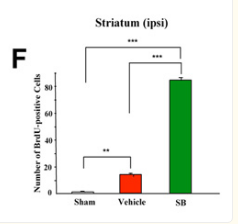
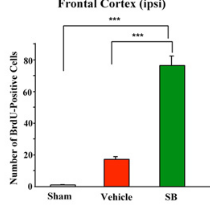
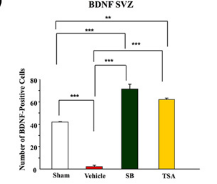
Day 14 after injury
-
@cs3000 said:
Looked into the effect of sodium butyrate on causing senescence / stopping cells proliferate. it can do that to normal fibroblasts (even tho showed increased wound healing). but effect is selective
I can't remember which study and which HDACi substance but the other day I also read a paper on wound healing and how, in vitro, almost every single crucial stage of cell line and activity suggested impairments of the different stages of wound closure - which overall however didn't materialize as wound healing was improved and "more structured".
I'm still wondering on what to stack or balance HDACi with to also promote proliferation of progenitor cells, though. Maybe aKG? Which may also induce OCT4, SOX2, KLF2 for reversal to pluripontent stem cells (iPSC)? -
In Ray Peat's newsletter "Epigenetics, sickness-aging, and changing science" (2014 01), which IMO starts off as a rather tiring albeit not unjustified rant, HDACi emerge as a significant factor against endometriosis and excessive menstruational symptoms (page 5):
- Although every organ seems to be renewed
(more or less regularly) by the maturation of stem
cells, the lining of the uterus is interesting because
it has a monthly cycle, in which stem cells multi-
ply actively during part of the month, and then
enter a phase of maturation, with the ability to
support a pregnancy. In the proliferative phase,
cells are dominated estrogen, and in the
maturation phase, by progesterone. During the
estrogen phase genes are massively silenced by the
HDAC enzymes, and during the progesterone
phase, those HDAC enzymes are inhibited.
Prolonged excessive exposure to estrogen with
deficient influence of progesterone can lead to the
development of endometriosis and endometrial
cancer. The combination of progesterone and
other HDAC inhibitors is effective in treating
endometriosis and endometrial cancer, and proba-
bly in other tumors, such as neuroblastomas (Atif,
et aI., 2011) and lymphomas. Cyproheptadine,
known mainly as an antiserotonergic antihista-
mine, is also an HDAC inhibitor, helpful in
treating mantle cell lymphoma (Paoluzzi, et al.,
2009). Aspirin, with an acetylating effect that has
been considered to be harmful, appears to activate
a histone, synergizing with a variety of HDAC
inhibitors to improve the effectiveness of cancer
treatments.
And for some more Ray Peat blessing fairy dust to the sake of this thread:
-
An epigenetic point of view suggests that a
generalized view of beneficial synergistic effects
should be considered--things that fundamentally
support the organism's full development activate
genes, and are antagonistic to things which funda-
mentally interfere with that development. Under
harmful conditions, genes are being silenced in
the organism's defense, in an organized way, and
b understanding the nature of that organisation,
more coherent interventions to protect the organ-
ism will be possible.
Radiation, heavy metals, hypoxia, and estro-
gen excess tend to create excess gene silencing,
and what they have in common is the creation of
over-excitable electrons, a reductive and nucleo-
philic state. The opposing' electronic state,
electron-withdrawing, typically with one or more
ketone groups in resonance with one or more
double bonds as in naphthoquinones (e.g., Inks, et
aI., 2012) characterizes many of the protective
substances, for example emodin (found in cascara
and aloe), curcumin, progesterone, caffeine and
theophylline.
Some chemicals known to have protective effects on
oxidative metabolism, such as short chain
saturated fatty acids and procaine and procaina-
mide, are also HDAC inhibitors with a broad
range of protective effects. Niacinamide (vitamin
B3) is a powerful HDAC inhibitor; vitamins A
and D have some synergistic interactions with
HDAC inhibitors. -
An optimal air pressure, or balance between
oxygen and carbon dioxide, regular exposure to
bright light, and foods that supply an appropriate
balance of amino acids, minerals, vitamins,
glucose and protective substances, such as HDAC
inhibitors, would help to support developmental
plasticity.
There we have Ray Peat endorsement of HDACis.
And the crucial caveat as to why this will remain in the fringes and shadows of "science":- Many commonly used drugs have unexpected
harmful epigenetic (degenerative) side-effects.
Csoka and Szyf have said (2009) "We propose
that epigenetic side-effects of pharmaceuticals
may be involved in the etiology of heart disease,
cancer, neurological and cognitive disorders,
obesity, diabetes, infertility, and sexual dysfunc-
tion." Although many researchers are now inter-
ested in an epigenetic approach, there is no
assurance that the medical and pharmaceutical
industry will ever make the adjustment, because
the most basic assumptions of their science are
challenged.
Ray Peat's Newsletter "Imprinting and Aging" (2015 07):
- In aging, expression of genes is broadly inhib-
ited by a general increase in DNA methylation.
Because of the involvement of similar epigenetic
modifications of gene expression, aging can be
thought of as a type of imprinting that involves
features similar to learned helpless[ness], in which the
organs and tissues, including the brain, are unable
to mobilize the energy needed for ordinary
adaptive processes.
He then mentioned vasopressin, which may be worth a good look on it being influenced by inhibitors of HDAC, DNMT, HMT. He reports that vasopressin also inhibits klotho and increases tissue calfication by increasing phosphate uptake and decreasing klotho-dependent renal phosphate clearance.
He suggested that such vasopressin along with microvesicles or exosomes of stressed cells is being being carried and released systemically [and from human to human, as is actually known], affecting all other cells and tissues of the body, dragging them into the same downturn.- Stress affects gene methylation to increase production of vasopressin. Antagonists to vasopressin can reverse the learned helplessness produced by stress. Vasopressin is responsible for the intestinal bleeding of stress, causing constriction of the bowel blood vessels, and damages the barrier function of the bowel, and also the blood brain barrier and generally increases vascular leakiness.
Here is why he maybe didn't warm up to SCFAs like butyrate, as he connected their synthesis to the many drawbacks associated with the abundance of gastrointestinal bacterial fermentation:
- The food industry is promoting the use of
various gums and starches, which are convenient
thickeners and stabilizers for increasing shelf-life,
with the argument that the butyric acid produced
when they are fermented by intestinal bacteria is
protective. However, intestinal fermentation
increases systemic and brain serotonin, and the
short-chain fatty acids can produce a variety of
inflammatory and cytotoxic effects.
- Although every organ seems to be renewed
-
@CrumblingCookie thanks for sharing. Great read.
Hello! It looks like you're interested in this conversation, but you don't have an account yet.
Getting fed up of having to scroll through the same posts each visit? When you register for an account, you'll always come back to exactly where you were before, and choose to be notified of new replies (either via email, or push notification). You'll also be able to save bookmarks and upvote posts to show your appreciation to other community members.
With your input, this post could be even better 💗
Register Login