@CO3 I’ve noticed amoxicillin is much tastier than erythromycin
stag
@stag
"A HERO, IN HIS MIND, WAS NOT SOMEONE WHO SUFFERED DISASTER AFTER DISASTER, HEROICALLY PULLING THROUGH WITH GREAT ENDURANCE, BUT RATHER ONE WHO FOCUSED HIS INTELLIGENCE AND SKILLS TO AVOID DISASTER, THUS SUCCEEDING BY GOOD PLANNING AND CRAFTY DECISION MAKING."
Latest posts made by stag
-
RE: anyone here take erithromycinposted in Not Medical Advice
-
Pure T3 Thyroid and Stories of Recovery from Chronic Fatigue Syndrome (ME/CFS) and Fibromyalgia: An Overview.posted in Literature Review
there are in fact three types of deiodinase enzymes that convert T4: D1 is responsible for conversion of T4 into T3 in most cells of the body, except for in the brain and the pituitary where D2 does the job (De Groot, 2015). D3 converts T4 into an inactive “reverse T3” (RT3) which (sometimes disturbingly) competes with T3 for space on the above-mentioned cells’ nuclear membrane thyroid receptors (note: D3 also converts T3 into T2; the role of this thyroid hormone is still being researched).
Critically, under certain conditions (such as inflammation, fasting or stress), D1 is downregulated and D3 is upregulated, resulting in less T3 and more RT3 at the cellular level. This can result in hypothyroid symptoms – even if the thyroid gland itself is functioning normally (Holtorf, 2014a).
the fact that some patients may have difficulty converting T4 into T3, that supplemented T4 may be converted into RT3 instead of T3, and that the transport of T4 into the cells requires more energy than the transport of T3 – may explain why therapies that contain T3 have often succeeded when T4-only therapies have failed to improve symptoms.

-
RE: Zonulin and its Consequencesposted in Bioenergetics Discussion
@LucH That's a very comprehensive paper, thanks for sharing!
I didn't mean that zonulin is a protective inhibitor, I was referencing that substances that inhibit zonulin (like Larazotide acetate) are protective against its deleterious effects (like type 1 diabetes / celiac), as elaborated on in the papers we have both posted.
@LucH said in Zonulin and its Consequences:
For me zonulin is secreted in presence of excess lectins and gliadin (agglutinin family) to avoid aggregation with L-glutamine from the membranes. Zonulin acts as a garde-barrière, telling the body to let the toxins get away. Zonulin tells the tight junctions to stay open …
Do you have more info on this? A search for lectins and leaky gut discloses this article:
Lectins: The Gluten-Lectin-Leaky Gut ConnectionSome lectins that we consume in everyday foods can bind to the sugars in the cell walls of the gut or in the blood. This can cause an immune response, leading to inflammation, intestinal damage, altered gut flora, malabsorption, decreased cellular repair, cellular death, and eventually disease.
These lectins bind to glycoproteins and glycolipids (sugar-coated proteins and fats) found on the surface of human and other animal cells. This binding allows for agglutination (clumping) and sometimes can produce an immune response. They can cause agglutination of blood cells and they can bind to the cells that line the small intestine.
This article also references Fasano, he seems to be a popular guy in zonulin world. The relevant reference regarding lectins is here (I think):
Dietary lectins are metabolic signals for the gut and modulate immune and hormone functionsA related paper is here:
Characteristics and consequences of interactions of lectins with the intestinal mucosaFollowing general Peat diet suggestions will have you avoiding most lectin-containing foods anyway, but there are two that stick out: dairy and nightshades. Someone could be getting most of their calories from milk and potatoes thinking theyre fine because its Peaty but may be driving intestinal permeability due to the lectin content.
The example given in the article of a noxious lectin is wheat germ agglutinin. I wonder how bad non-wheat derived lectins like those from potatoes and milk are.
If the mechanism is as described I also wonder if it may be advisable to keep dietary gluten and lectins to a minimum during L-glutamine supplementation, as that combination may provide ample reason for a zonulin trigger. This will be easy for wheat, likely also potatoes, but cutting milk may prove to be a challenge, if necessary. It also may be the case that the supraphysiological doses of glutamine generally used in supplementation may override any zonulin signaling caused by incidental dietary lectins. I'm not sure.
-
Zonulin and its Consequencesposted in Bioenergetics Discussion
Tight Junction(TJ) proteins are crucial components of the cellular structures that form barriers between cells in epithelial and endothelial tissues. These proteins help maintain cell polarity and control the passage of molecules and ions through the space between cells, known as the paracellular pathway. Dysfunction of tight junctions is casually referred to as leaky gut.
Leaky Gut and Autoimmune Diseases*
A century ago, TJs were conceptualized as a secreted extracellular cement forming an absolute and unregulated barrier within the paracellular space [22]. Biological studies of the past several decades have shown that TJs are dynamic structures subjected to structural changes that dictate their functional status under a variety of developmental scenarios.
The discovery of zonula occludens toxin (Zot), an enterotoxin elaborated by Vibrio cholerae that reversibly opens TJ[23], increased our understanding of the intricate mechanisms that regulate the intestinal epithelial paracellular pathway and led to the discovery of its eukaryotic counterpart zonulin [24, 25].
This pathway appears to be involved in several functions, including TJ regulation responsible for the movement of fluid, macromolecules, and leukocytes between the bloodstream and the intestinal lumen and vice versa.
GI symptoms in diabetes mellitus have been generally ascribed to altered intestinal motility secondary to autonomic neuropathy [35].However, other studies suggest that an increased permeability of intestinal TJ is responsible for both the onset of the disease and the GI symptoms that these patients often experience [36]. This hypothesis is supported by a study performed on a spontaneously diabetic animal model [37]. The authors of this study showed an increased permeability of the small intestine of Bio Breeding Diabetes Prone (BBDP)/Wor diabetic-prone rats that precedes at least a month the onset of diabetes [...] We confirmed these data by reporting in the same rat model that zonulin-dependent increase in intestinal permeability precedes the onset of T1D by 2–3 weeks [38]. Oral administration of the zonulin inhibitor, AT1001 (now called Larazotide acetate), to BBDP rats blocked autoantibody formation and zonulin-induced increases in intestinal permeability, so reducing the incidence of diabetes [38]. These studies suggest that the zonulin dependent loss of intestinal barrier function is one of the initial steps in the pathogenesis of T1D in the BBDP animal model of the disease. The involvement of zonulin in T1D pathogenesis was corroborated by our studies in humans showing that ~50% of T1D patients has elevated serum zonulin levels that correlated with increased intestinal permeability [39].
In clinically asymptomatic Crohn’s disease patients, increased intestinal epithelial permeability precedes clinical relapse by as much as 1 year, suggesting that a permeability defect is an early event in disease exacerbation
Before you get too excited, read this sensible article positing that serum zonulin isnt a reliable biomarker to test for leaky gut: The Clinical Utility of Zonulin Testing
here's a tidbit:
The basic idea is that with zonulin, a fairly large (47 kDa) protein produced in the gut, should not be able to pass into the bloodstream under normal conditions. Thus, its recovery in plasma or serum likely reflects the degree of intestinal permeability. While the theory makes sense, any good biomarker worth its predictive value will also have the following characteristics:
- Sensitivity
- Specificity
- Robustness
- Accuracy
- Reproducibility
In the next several sections, I’ll discuss why zonulin doesn’t hold up to these standards.
Note his final section, "Is It Even Worth Testing for Leaky Gut?" is ill-informed, asserting:
intestinal permeability itself is almost always caused by something further upstream
this is incorrect, as the original article demonstrates:
1. Elevated serum zonulin precedes symptoms
2. Zonulin inhibitors are protectiveHowever there are still a handful of reasonable contentions with looking only at zonulin, and some actionable alternatives provided.
Hopefully this is instructive to those who feel they may be suffering from leaky gut/autoimmune conditions and are looking for specific mechanisms of action and practical interventions.
*full text available in the usual places
-
Progesterone is protective against lethal doses of histamine and endotoxin, may bind to endotoxin directlyposted in Bioenergetics Discussion
haidut made an underrated post on The Forum That Shall Not Be Named on the protective effect of progesterone on cats administered lethal doses of histamine and endotoxin. At the time of the post the study was not available anywhere online. I am pleased to announce the full text is now indexed by scihub, available here:
Most notable is the possibility that progesterone may actually bind to and inactivate endotoxin directly:
Endotoxin, progesterone, and endotoxin that had been incubated at 37°C. with progesterone for one-half hour were extracted with ether. The ether was evaporated and the resulting extract diluted with saline. Progesterone is ether-soluble, and as was expected, when the progesterone was extracted by itself most of the progesterone was in the ether extract. When ether extracts of the progesterone-endotoxin mixture were prepared, there was little yield in the ether extract. The ether and aqueous extracts of endotoxin and the endotoxin-progesterone mixture were administered to a series of animals and the blood pressure and survival rate determined.
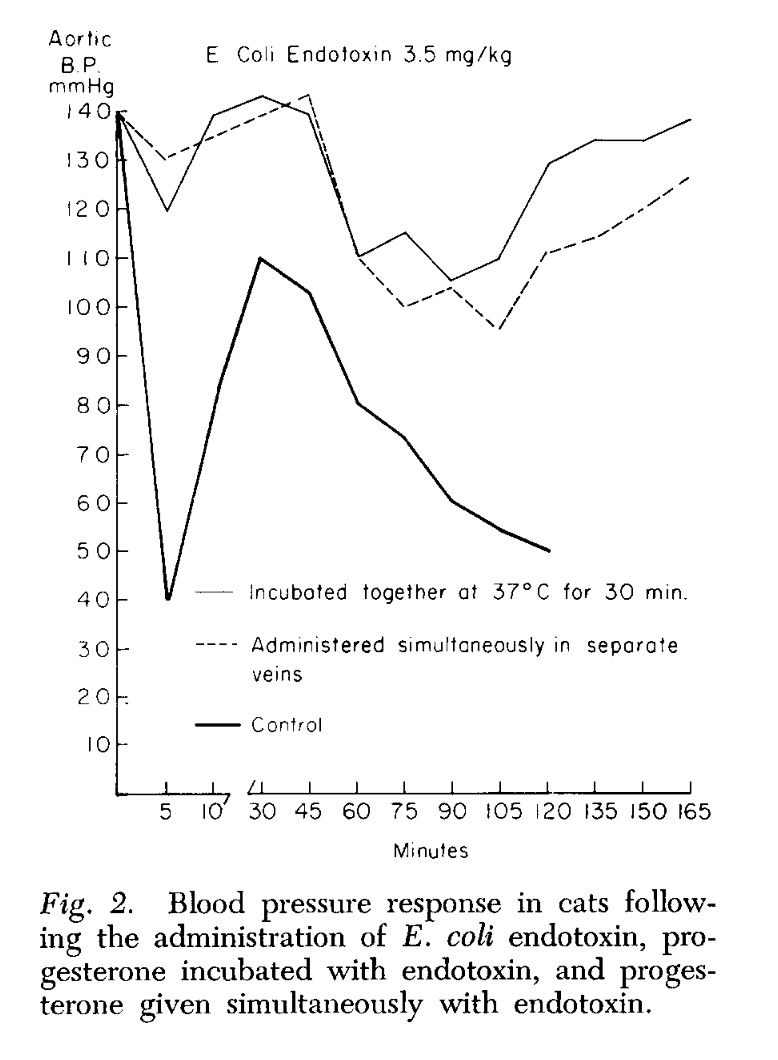
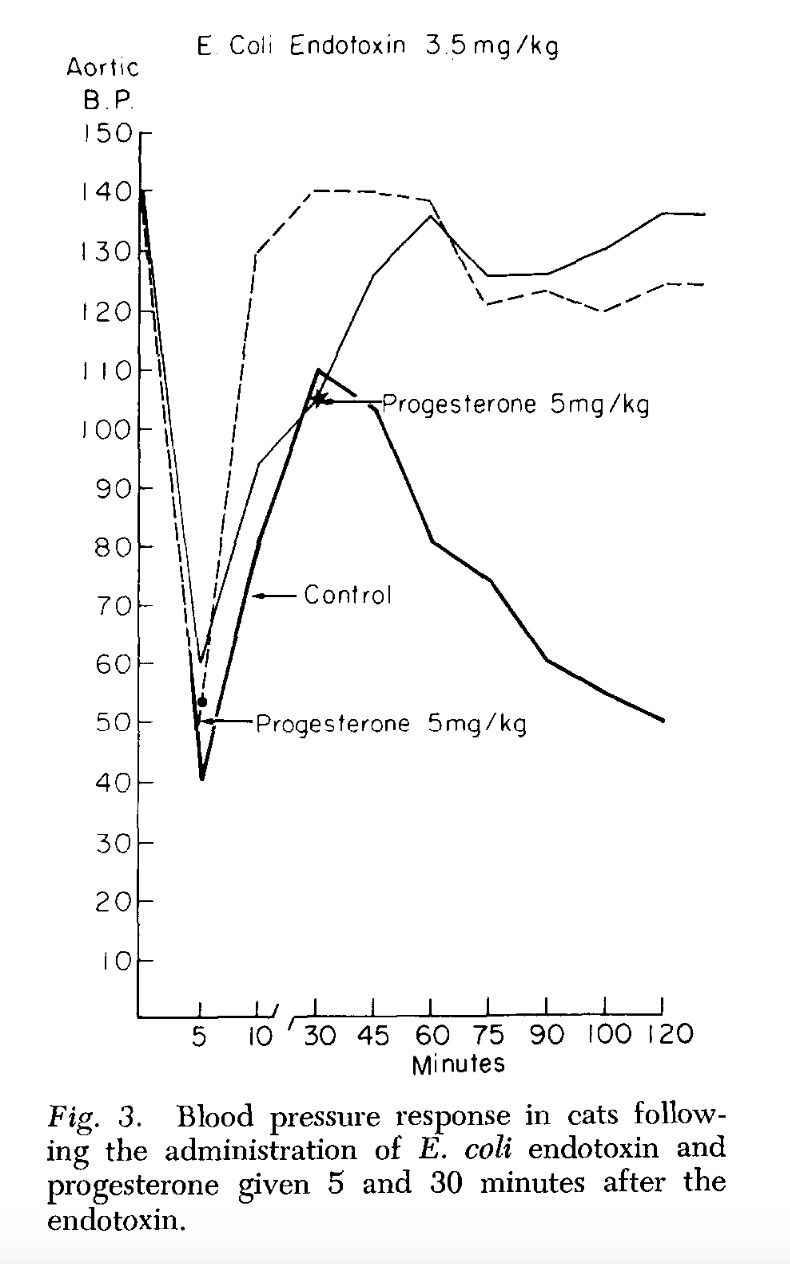
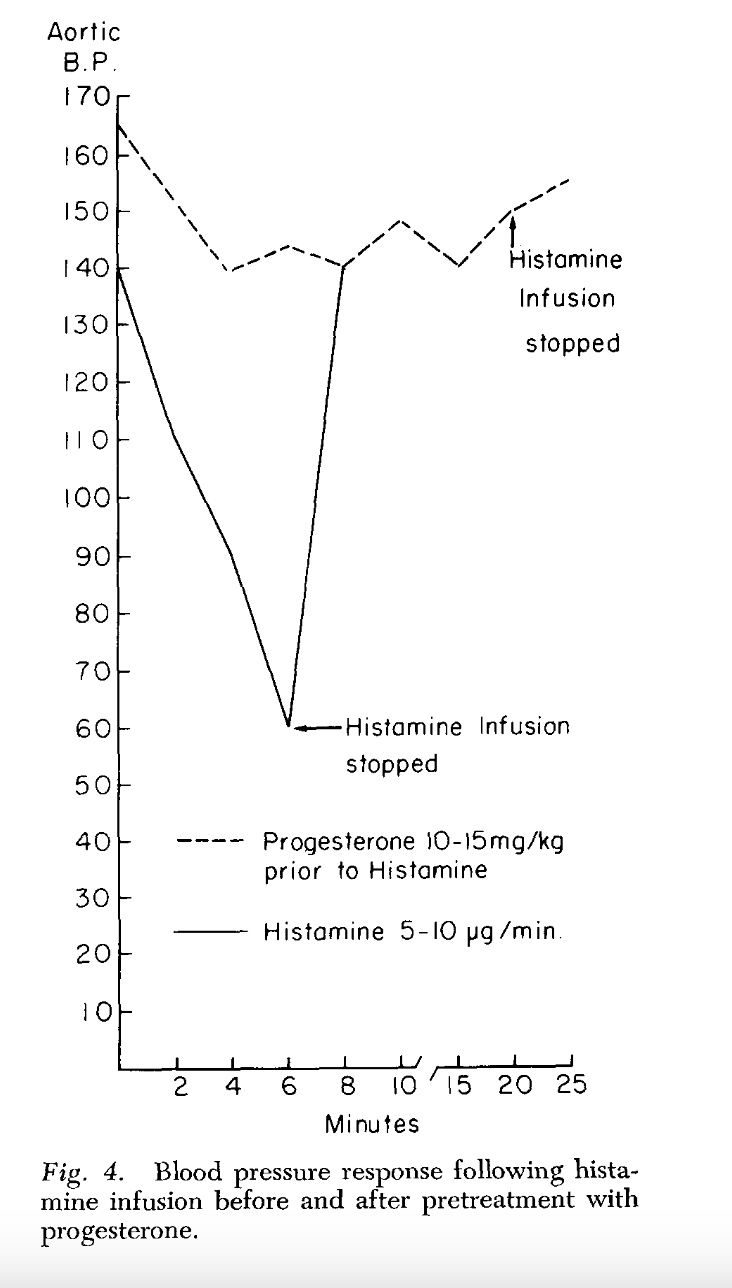
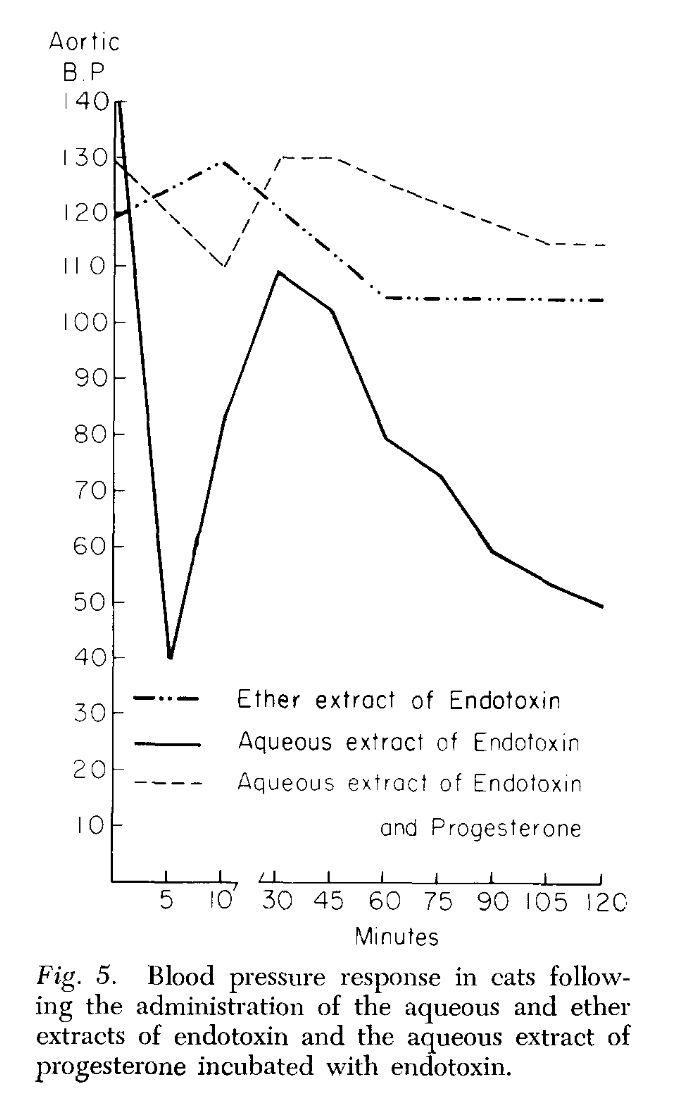
The preliminary results of our work with the ether and aqueous extracts of progesterone and endotoxin, and the increased survival obtained with prior incubation of endotoxin with progesterone, appear to indicate that progesterone may form an in vitro bond with the endotoxin compound. This has been suggested as a possible alternate explanation of the action of aldosterone and endotoxin (3, 4). Whether this results in direct inhibition of the endotoxin or the inhibition is mediated through unknown substances in vivo is pure conjecture at this time.
HED is 1mg/kg, or ~80mg for a ~180lb subject. I can personally attest that this dosage is far more effective than activated charcoal or any other binder when it comes to alleviating heavy endotoxin load.
haidut's original blogpost can be found here: Progesterone may bind and deactivate endotoxin (LPS) directly
-
Antimicrobial Activity of Spices Popularly Used in Mexico against Urinary Tract Infectionsposted in Literature Review
Free article, nice review of the "hot oils" and their antimicrobial capacity:
Antimicrobial Activity of Spices Popularly Used in Mexico against Urinary Tract Infections
Abstract
Urinary tract infections (UTIs) are the most common infectious diseases worldwide. These infections are common in all people; however, they are more prevalent in women than in men. The main microorganism that causes 80–90% of UTIs is Escherichia coli. However, other bacteria such as Staphylococcus aureus, Enterococcus faecalis, Pseudomonas aeruginosa, Proteus mirabilis, and Klebsiella pneumoniae cause UTIs, and antibiotics are required to treat them. However, UTI treatment can be complicated by antibiotic resistance and biofilm formation. Therefore, medicinal plants, such as spices generally added to foods, can be a therapeutic alternative due to the variety of phytochemicals such as polyphenols, saponins, alkaloids, and terpenes present in their extracts that exert antimicrobial activity. Essential oils extracted from spices have been used to demonstrate their antimicrobial efficacy against strains of pathogens isolated from UTI patients and their synergistic effect with antibiotics. This article summarizes relevant findings on the antimicrobial activity of cinnamon, clove, cumin, oregano, pepper, and rosemary, spices popularly used in Mexico against the uropathogens responsible for UTIs.
-
Incident report: SIBO, molybdenum, gout, and aspirinposted in Experimental Logs
An overgrowth of sulfate-reducing bacteria produces a lot of hydrogen sulfide gas. Some of that gas is farted out, but up to 70% is diffused through the tissues. That sulfide must be detoxified through the sulfide -> sulfite -> sulfate pathway.
The jump from sulfite -> sulfate requires the enzyme sulfite oxidase (SUOX). suox requires the trace mineral molybdenum to function. I was taking up to 1mg molybdenum to support suox.
Molybdenum is also a cofactor for xanthine oxidase, the enzyme that facilitates the creation of uric acid, which can precipitate urate crystals and cause gout. I had been megadosing Mo (500-1000mcg for ~1wk, plus lower doses off and on for a month prior) and went for a 5mi walk and accidentally gave myself a gout flare in my feet.
Allopurinol is prescribed for gout, as it is a xanthine oxidase inhibitor, but that only stops the formation of new uric acid. Once the uric acid has been created, it must be excreted via urine.
With no prescriptions immediately available but the pain necessitating a timely response, I attempted to replicate a protocol¹ where aspirin had a biphasic effect on urinary urate excretion, with a big increase at high doses (3g+). My previous highest aspirin dose was 1.5g.
Aspirin will need to be paired with other ancillaries for safety/efficacy. 325:1 is the rule of thumb for aspirin:k2. Glyprin pills use a 2:1 glycine:aspirin ratio so thats a good ballpark. I use perfect brand gelatin which contains 2.6g glycine in one scoop, so a 2.6:1 ratio is simpler for me.
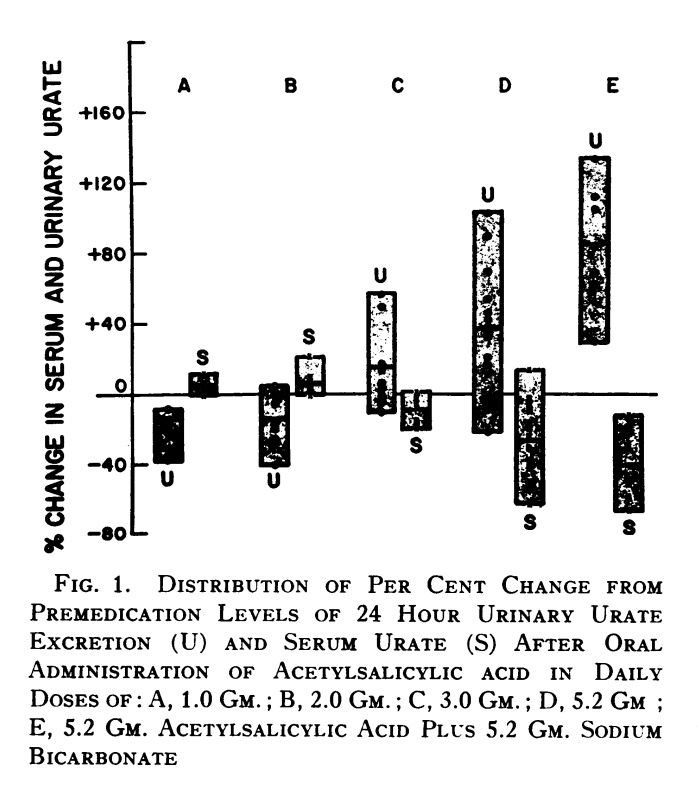
In addition to demonstrating how the uricosuric effect of aspirin starts at 3g, this table also demonstrates a 1:1 ratio of baking soda:aspirin increased urinary urate clearance more than aspirin alone (column E)With these ratios established, I used an electric kettle to heat filtered water up to 140F and mixed in:
1g aspirin
3mg k2
2.6g glycine via gelatin
1g baking soda
honey to tastewith the intention to do this 5 times, once per hour, until 5g aspirin goal is reached. This is replicating the study, which divided the 5.2g aspirin dose into multiple doses over the course of a day. I also took 500mg B1 once at the beginning.
I had a meal of liver and rice during the dosage period.
The first two doses went well, but after my third dose, I became very fatigued, dizzy, and sensitive to light, with tingling tongue and lips. These symptoms were remedied by eating several tablespoons of honey straight from the jar to taste.
The assumption here is that both liver and aspirin combined to drastically lower my blood sugar past what I had been replenishing it with. Having drastically underestimated the calorie/carb requirements of high dose aspirin, I called off the experiment, ate, and went to bed.
I also experienced tinnitus while falling asleep, which disappeared after ~7hrs of deep restful sleep. I did not experience any bleeding or ulcers.
This reaction makes sense, as at around 3g aspirin becomes a mitochondrial uncoupler that would necessitate higher nutrient requirements.
Despite having to call it off early, I did manage to reach the 3g threshold for improved urate excretion, and my gout symptoms did largely disappear, as well a noticeable improvement in general inflammatory symptoms.
Peat has brought up the idea that gout is actually caused by endotoxin-coated phosphate crystals. Seneff has also proposed the glyphosate plays a role in gout flares.²
These ideas probably have merit, but I think the fact that low-dose aspirin can induce a gout flare³⁴⁵⁶⁷ while a high dose can remedy it implies uric acid plays at least some significant role, given aspirin's dose-dependent effect on urate excretion.
High dose aspirin could work as a remedy for gout flares, but it requires some prep and intention to do right, notably a reliable carb source. It's worth noting that acidic urine will stop uric acid excretion so orange juice is not a good choice for this protocol. The original study is very interesting, and worth the read.
- STUDY OF THE PARADOXICAL EFFECTS OF SALICYLATE IN LOW, INTERMEDIATE AND HIGH DOSAGE ON THE RENAL MECHANISMS FOR EXCRETION OF URATE IN MAN
- Can glyphosate’s disruption of the gut microbiome and induction of sulfate deficiency explain the epidemic in gout and associated diseases in the industrialized world?
- Association Between Low-Dose Aspirin and Uric Acid in the Elderly: An Observational Retrospective Cross-Sectional Study
- Low-dose aspirin use and recurrent gout attacks
- The effect of mini-dose aspirin on renal function and uric acid handling in elderly patients
- The Effect of Low Dose Aspirin on Serum and Urinary Uric Acid Level in Gouty Arthritis Patients
- Acute attack of gout precipitated by concomitant use of aspirin and diuretic in a rheumatic mitral stenosis patient
- fwiw low dose aspirin only seems to instigate gout flares in people already predisposed to gout, and one of the studies found no correlation
-
RE: Low-dose naltrexone for IBS/SIBOposted in Literature Review
References
- About Low Dose Naltrexone
- Low-Dose Naltrexone (LDN)—Review of Therapeutic Utilization
- Low-dose naltrexone for disease prevention and quality of life
- Complex Presentations, Identification and Treatment of Mast Cell Activation Syndrome and Associated Conditions: A Case Report
- Low Dose Naltrexone: Side Effects and Efficacy in Gastrointestinal Disorders
- Longest Levers: LDN - @anabology
-
Low-dose naltrexone for IBS/SIBOposted in Literature Review
Literature Review
A selection of LDN articles, mostly as it relates to GI conditions like IBS and SIBO. I have highlighted certain parts of the excerpts and organized the order in which they appear in hopes of streamlining continuity of ideas.
Introduction
The drug naltrexone was approved by the FDA in 1984 in a 50mg dose for the purpose of helping patients with heroin or opium addiction, by inhibiting the effect of such drugs. By blocking opioid receptors, naltrexone also blocks the reception of the opioid hormones that our brain and adrenal glands produce: beta-endorphin and metenkephalin. Many body tissues have receptors for these endorphins and enkephalins, including virtually every cell of the body's immune system.
In 1981, Dr. Patricia McLaughlin and Dr. Ian Zagon of Penn State began researching and later published results on the use of small doses of naltrexone to retard tumor growth in animals.
In 1985, Bernard Bihari, MD, a physician with a clinical practice in New York City, discovered the effects of a low dose of naltrexone (approximately 3mg once a day) on the immune system of the human body. He found that this low dose, taken at bedtime, was able to enhance a patient's response to infection by HIV, the virus that causes AIDS. (Subsequently, he found the optimal adult dosage of LDN to be 4.5mg.)
In the mid-1990's, Dr. Bihari saw that patients in his practice with cancer (such as lymphoma or pancreatic cancer) could benefit, in some cases dramatically, from LDN. In addition, people who had an autoimmune disease (such as lupus) often showed prompt control of disease activity while taking LDN.
Low-Dose Naltrexone (LDN)—Review of Therapeutic Utilization
Naltrexone or 17-(cyclopropylmethyl)-4,5-epoxy-3,14-dihydroxymorphinan-6-one is a non-selective pure opioid antagonist with the highest affinity for μ-opioid receptors [6,9]. It is almost completely absorbed (96%), but its oral bioavailability ranges between 5% and 40% due to first-pass metabolism. Naltrexone’s half-life is 4 h and it is a highly metabolized (>98%) drug—the major metabolite being 6-β-naltrexol with a half-life of 13 h and antagonist action on opioid receptors. Glomerular filtration is the predominant mode of renal elimination for a small fraction of unmetabolized naltrexone, while 6-β-naltrexol is additionally secreted.
Mechanism of Action
Rebound Effect
Low-dose naltrexone for disease prevention and quality of life
Naltrexone, an orally effective, long-lasting opiate receptor antagonist, was approved by the FDA for treating alcohol and opiate addiction in 1984, but its general patent expired the following year. It is a non-selective antagonist, with robust effects on pleasure promoting mu opioid receptors (MOR) and delta opioid receptors (DOR) [1], with less antagonism of aversion-mediating kappa opioid receptors (KOR) [2] but substantial effect on the more recently discovered orphanin FQ or nociceptin [N/OFQ] opioid family [3]. The benefits of high dose naltrexone in narcotic addiction are explained by blockade of all pleasure producing effects of opioids, and similar mechanisms may explain the ability of naltrexone to reduce binging on alcohol.
The normal 50 mg naltrexone dose that blocks opioid receptors 24 h per day is commonly prescribed for alcoholics and heroin addicts who wish to resist a relapse. This typically amounts to more than 0.5 mg/kg for most adults. In contrast, the most common LDN use is typically 4.5 mg, which generally means most adults get no more than 0.08 mg/kg per day, which can block mu opioid receptors for only a few hours, perhaps up to 6 h. If taken at bedtime, this would mean that an individual might wake up the next morning with a homeostatic rebound-induced over-activity of their own endogenous opioid systems.
When administered in low-doses of 3–4.5 mg daily, naltrexone increases the expression of mu, delta, and epsilon opioid receptors as well as central and circulating met-enkephalin (ME) and betaendorphin (BE)
Zagon and McLaughlin concluded that LDN increased opiate receptors and elevated circulating BE and ME after a 4–6 h period of receptor blockade. This ‘‘rebound phase” may release the increased density of mu and delta opioid receptors for endogenous opioid stimulation with the increasingly available BE and ME. The general principle operative here may be that the increased concentrations of BE and ME that gain access to increased density of MOR and DOR receptors may ‘‘functionally supersensitize” [24] endogenous opioid functions throughout the body with beneficial downstream effects on various body parameters, especially immunocompetence.
Recent work on collagen-induced arthritis in rats have found BE treatment to reduce clinical arthritis manifestations by shifting the balance of TH-1 and TH-2 cells toward TH-2. This comes from down-regulating the NF-kappa2 pathway, including tumor necrosis factor alpha, Interleukin-1beta, Interleukin-6, inducible nitric oxide synthase, and mRNA for matrix metalloproteinase-2 and mmp-9 [32]. Dr. Sacerdote and her colleagues in Milan have reached the same conclusion that BE increases ameliorate autoimmune diseases by suppressing TH-1 and augmenting TH-2 cells [33].
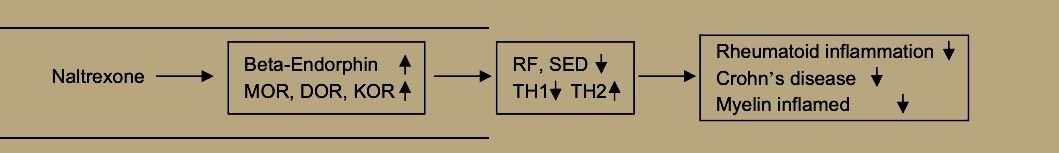
Plotnikoff et al. [34] report that ME stimulates expression of interleukin-2 receptors and blood levels of interleukin-2, along with increases in white blood cells, natural killer cell activity, gamma-interferon, active T-cells and other elements of the immune system
Both ME and BE may enhance NK cell activity via the mu receptor [38] and also by binding to receptors on cancer cells themselves [23].
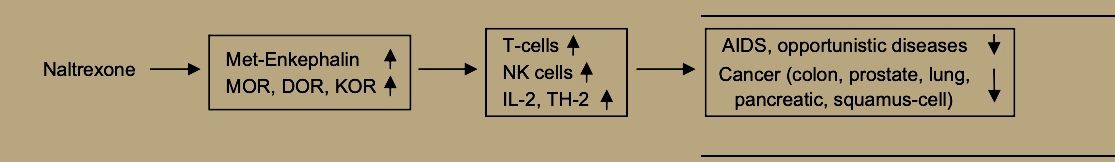
Thus, LDN, through its enhancement of immune functions [21] and specifically of natural killer cell activity [49] may promote prevention and treatment of viral diseases and bacterial infections. Evidence from animal models suggests that naltrexone’s path to supporting immune defenses against viral disease begins by increasing both beta-endorphin and met-enkephalin, which may then bind to sensitized mu opioid receptors to increase natural killer cell activity for quelling viral infection [50].
OGFr Antagonism
OGFr: This is the opioid growth factor receptor, and naltrexone blocks opioid growth factor from binding to it. Despite OGF being called a growth factor, it inhibits regenerative growth. Blocking the OGFr can promote repair, but only during naltrexone binding. You likely don't want permanent growth, repair, or cell division, so it's good that LDN is cleared out relatively quickly. You get a window of repair, and then revert to baseline. It's very tunable and generally safe. The anti-cancer effects of LDN are likely a product of the phase where naltrexone is not bound to the receptor, for example, but blocking the receptor does not give you cancer. This will give us some insight into the protocol below.
Low-Dose Naltrexone (LDN)—Review of Therapeutic Utilization
The upregulation of the endogenous opioid system is evident in experimental models by rising levels of endorphin and met-enkephalin, also known as opioid growth factor, with concomitant respectively increased μ-opioid, δ-opioid, and ζ-opioid receptor expression, the latter also termed opioid growth factor receptor [24,26,27,28,29].
The higher reactivity of immune cells and decreased growth of cancerous cells are both mediated by transient increase in opioid growth factor signaling [23,24,30,31,32]. Permanent blockade of opioid growth factor receptor leads to enhanced cellular growth, which is unwanted in case of tumors, but has been experimentally used for wound or corneal abrasion healing (for a comprehensive review on findings and mechanisms see [33]).
Experimental studies in mice with induced autoimmune encephalomyelitis, a standard MS model, demonstrated evidence of opioid growth factor signaling as a salient feature in pathophysiology of MS. Prior to any clinical symptoms expected in mice, there was a reduction in circulating opioid growth factor [51]. After introduction of LDN therapy, the values of opioid growth factor were restored. Previous in vitro experiments reported that opioid growth factor or LDN may suppress proliferating B and T cells [52,53], a feature with implications for autoimmune states. A recent experiment, based on the aforementioned MS mouse model, demonstrated that opioid growth factor or LDN therapy decreased levels of interferon-γ, TNF-α, and IL-10, but increased those of IL-6 [54]. This could possibly help achieve a response state favoring a Th2 immune profile, otherwise considered to ameliorate MS [54]. An earlier study in the same mouse model demonstrated benefits of opioid growth factor and LDN in terms of halting disease progression, reversing neurological deficits, and considerably delaying onset of neurological dysfunction [24]. Reduced serum opioid growth factor levels were also noticed in humans affected by MS and likewise did LDN therapy revert the discrepancy.
TLR-4 Antagonism
TLR4: This is the receptor for 'endotoxin.' Endotoxin is a component of bacterial cell walls that triggers an inflammatory reaction. The extreme of endotoxin poisoning is known as sepsis. If you have a leaky gut, or have rapid growth and turnover of bacteria in your gut (SIBO, for example, or if you eat highly fermentable fibers), endotoxin is likely leaking into your blood with no real threat of infection. This was not as common in ancestral times due to higher quality foods, so your body is right to overreact. Now, though, you don't want your body to overreact, and LDN can be a much-needed bandage as you sort out gut issues. It blocks TLR4 and prevents the endotoxin-mediated inflammatory cascade.
Low-Dose Naltrexone (LDN)—Review of Therapeutic Utilization
In discrete ‘low-doses’ ranging from 1 to 5 mg, naltrexone acts as a glial modulator [14,15]. It specifically binds to Toll-like receptor 4, where it acts as an antagonist [8,16,17]. Toll-like receptor 4 downstream cellular signaling includes myeloid differentiation primary response 88 (MyD88) and toll-interleukin receptor (TIR)-domain-containing adapter-inducing interferon-β (TRIF) pathways, both ultimately leading to inflammatory end-products such as interleukin (IL)-1, tumor necrosis factor (TNF)-α, interferon-β, and nitric oxide [18]. Low-dose naltrexone disrupts the TRIF portion of the signaling cascade which reduces TNF-α and interferon-β synthesis [8]. Consequently, activated microglial cells expressing Toll-like receptor 4, otherwise a non-constitutive receptor, exert an attenuated pro-inflammatory profile [16].
A particular feature of stereoselectivity and LDN targets is noted. While classic opioid receptors are selective for (−)-opioid-isomers, Toll-like receptor 4 is not [8]. By utilizing (+)-naltrexone or (+)-naloxone, opioid related signaling would not be affected and Toll-like receptor 4 would be exclusively targeted.
Low-dose naltrexone is a mu-opioid antagonist using pharmacologically low doses (1 to 5 mg). The proposed mechanisms of LDN include acting as a glial modulator, inducing the production of endorphins, and binding to and antagonizing toll-like receptor 4, which eventually produces inflammatory products such as tumor necrosis factor(TNF)-α, interferon-β, interleukin (IL)-1, and nitric oxide.14 These inflammatory proteins are proposed to activate mast cell activity. Additionally, endorphins are associated with migrating motor complex (MMC) improvement and thus it can be theorized that LDN acts as a mild prokinetic. Opioid antagonists are also proposed to modify gastric motility by stimulating peristalsis and therefore increasing transit time.15 There are currently no clinical guidelines for the use of LDN. Clinical research to date consists mostly of the use of LDN as a promising treatment for chronic inflammatory pain conditions. Clinical trial abstracts currently show LDN being studied for use in a wide range of immune-mediated conditions, including regional complex pain syndrome, painful diabetic neuropathy, psoriasis, inflammatory bowel disease (IBD), chronic fatigue syndrome, autism, opioid side effects, prevention and treatment of immunothrombosis in Covid-19, relapsing depression, multiple sclerosis (MS), and fibromyalgia. There are currently 62 clinical trials studying LDN in as wide-ranging of conditions.16,17 The majority of research concerning gastrointestinal conditions is centered around its stimulation of mucosal healing in IBD, though it is often used off-label as a mild prokinetic agent in the management and relapse prevention of SIBO.14
Final outcomes include immense improvement upon mast cell stabilization with ketotifen, and remission of SIBO with low-dose naltrexone (LDN).
Selective Dual-mode Opioid Signaling Inhibition
This appears to only be relevant to ULDN not LDN but I have included it because the proposed relevance to IBS is interesting.
Low-Dose Naltrexone (LDN)—Review of Therapeutic UtilizationUltra low-dose naltrexone or naloxone (ULDN) pertains to a dosing range when less than 1 μg quantities of drug are used. Its mechanism of action is related to a bimodal cellular response to opioids. In addition to their inhibitory Gi-coupled response, opioids induce a concomitant and less overt Gs-coupled stimulatory response [35]. The stimulatory response is acutely exclusive if small quantities of opioid agonists are used, otherwise it increases steadily with chronic μ-opioid receptors stimulation. The opioid receptor Gs-coupled response cascade has been associated with prolongation of action potential, hyperalgesia, tolerance, and dependence. A crucial element mediating μ-opioid receptor second messaging is the scaffolding protein filament called filamin-A (FLNA) [36,37]. Filamin-A contains a high-affinity binding site for naloxone and naltrexone (3.94 pM). When such a binding occurs, μ-opioid receptor Gs-coupling is attenuated and Gi-coupled response prevails. Thus, analgesic effects of opioids are potentiated and unwanted consequences are mitigated. However, FLNA also contains a low-affinity binding site for aforementioned opioid antagonists (834 pM). If both binding sites are saturated, the favorable profile of μ-opioid receptor signaling is abolished. These affinity sites dictate the span in which ULDN may be clinically relevant in boosting responses to μ-opioid receptor agonism. Corresponding calculated drug concentration ranges are 1.3–272.9 pg/mL for naloxone and 1.4–284.7 pg/mL for naltrexone.
Low-Dose Naltreoxone for the Treatment of Irritable Bowel Syndrome: A Pilot Study
As a result of intensive study of the brain–gut axis over the last decade, there is general agreement that IBS patients suffer from visceral hypersensitivity [17] and are more sensitive than normal controls to bowel distension [18].
Patients with IBS appear to have altered sensitivity to exogenous opioids [11], probably due to altered central release of endogenous opioids in response to visceral stimulation.
Opioid agents have a marked effect on gut motility and secretion and can alleviate visceral pain [4]. An in vitro study in a mouse dorsal root ganglion (DRG) model demonstrated dual modulation of action potential duration. Micromolar opioid concentrations shorten action potential duration, decrease Ca2+ flux and neurotransmitter release, and produce an inhibitory or analgesic effect [5]. These effects are blocked by high-dose naltreoxone (NTX), an opioid antagonist [5]. Paradoxically, nanomolar opioid concentrations can prolong the action potential duration, increase Ca2+ flux and neurotransmitter release, and cause hyperalgesia rather than analgesia. These effects are blocked by low-dose NTX [5].
Crain and Shen characterized two opioid receptor systems that mediate functionally opposite effects [5–7]. The overall effect of opioid agonists depends on the dominant receptor population, the low-affinity population (at which high opioid concentrations are inhibitory), or the high-affinity population (at which low opioid concentrations are excitatory). The excitatory opioid response is selectively blocked by extremely low doses of opioid antagonists (such as NTX), but the inhibitory response is not. Consequently, low-dose NTX can enhance the analgesic potency of opioids [5]. In vitro studies in DRG neurons have shown that very low doses of NTX alone enhance opioid analgesia and attenuate the development of opioid tolerance and/or dependence [8].
The excitatory properties of opioids often go undetected in humans, probably because they are partially masked by the inhibitory analgesic effects at the usual clinical doses. Normally, the inhibitory pathway for opioids is the dominant pathway. However, in IBS patients there may be an altered central release of endogenous opioids in response to visceral stimulation [11], making the excitatory pathway predominant and resulting in pain.
The bimodal excitatory and inhibitory opioid receptor function that exists in sensory and DRG neurons was proved in vivo in a mouse model [9]. A bimodal excitatory and inhibitory opioid receptor function similar to sensory DRG neurons exists in myenteric neurons [10]. Thus, it is possible that the selective antagonism of excitatory opioid receptor function in DRG and myenteric neurons caused by low doses of opioid antagonist could enhance the analgesic effects of endogenous opioids on visceral sensory neurons and attenuate the hypersensitivity seen in visceral sensory and motor neurons in IBS patients [10].
The study drug was well tolerated and did not have significant side effects; there was no nausea or vomiting, which are associated with opiate antagonists. The results of the present study point to a positive response rate in IBS patients and support an analgesic effect, even controlling for a potential placebo effect of the study drug: about 76% of patients graded themselves as responders by the end of the treatment period. When patients were subgrouped by IBS phenotype, the alternating-type group had the highest response rate (86.7%), followed by the diarrhea-predominant group (75%) and then the constipation-predominant group (70%). The increase in the number of pain-free days was statistically significant (P = 0.011), more prominent in males, and lasted for 2 weeks after treatment termination.
The more prominent pain response in males is interesting and needs to be verified in further studies.
(this was worded poorly imo, I think they meant the more prominent pain-alleviating response)
Efficacy
Low Dose Naltrexone: Side Effects and Efficacy in Gastrointestinal Disorders
Naltrexone was used to supplement stable existing therapy in IBD patients or act as sole treatment in the cases of IBS-SIBO and idiopathic IBS. Patients with chronic constipation were treated with LDN alone or as an adjunct to other partially effective medications
Of the 13 patients with idiopathic IBS (3 with diarrhea, 5 with constipation, and 5 with alternating bowel habits). the results were:
- 2 (13.3%) were markedly improved
- 5 (38 5%) were moderately improved
- 2 (15.3%) were unchanged
- 3 (23.1%) were markedly worse
Of the 3 that were worse. the results were:
- 1 had IBS-constipation
- 2 had IBS-alternating bowel habits
The 85 patients with IBS-SIBO were treated for a mean of 14.2 weeks (58 with 2.5 mg and 27 with 2.3 mg twice daily LDN), with the following results:
- 15 (17.6%) were markedly improved
- 32 (37.6%) were moderately improved
- 11 (12 9%) were mildly improved
- 23 (27.0%) were unchanged
- 3 (3.5%) were moderately worse
- 1 (1 .2%) were markedly worse
A second course of antibiotics were administered in 38% of these patients during the 14 weeks to retreat recurrent symptoms of SIBO. LDN (2.5 mg twice daily) was administered for a mean of 10.8 weeks in 12 patients with chronic constipation. Of these patients, the results were:
- 7 (58.3%) were markedly improved
- 1 (8.3% was moderately improved
- 3 (20.0%) were mildly improved
- 1 (8.3%) was unchanged
Eight patients with inflammatory bowel disease (4 Crohn's and 4 ulcerative colitis) were treated with 4.5 mg naltrexone daily for a mean of 16.8 weeks, with the following results:
- Two were markedly improved
- 1 was moderately improved
- 1 was mildly improved
- 4 were unchanged
Two of those who stated they were unchanged were in clinical remission prior to starting
Low-Dose Naltrexone (LDN)—Review of Therapeutic Utilization
The first application of LDN in gastrointestinal-related issues was in 2006, when an Israeli research group presented a pilot study involving 42 patients suffering from irritable bowel syndrome (IBS) [59]. It was an open-label study where 0.5 mg LDN was given daily for 4 weeks. The drug was well tolerated and more than 75% of patients were considered responders per a subjective scale measuring pain-free days and symptom relief. Later on, a number of studies regarding inflammatory bowel disease (IBD) were conducted (Table 3).
One of the earliest was an open label study involving 17 patients with histologically active disease and Crohn’s disease activity index (CDAI) score of 220–450 [38]. Low-dose naltrexone was given in a 4.5 mg daily dose over a period of 12 weeks. After the treatment, 89% of the patients were deemed responders with a decrease in CDAI score by 70 points, while 67% achieved disease remission.
The most recent clinical study assessing LDN in IBD was a prospective open-label trial involving 28 patients affected by Crohn’s disease and 19 by ulcerative colitis [44]. Patients with an intractable active phase of IBD received 4.5 mg of LDN daily in addition to the standard treatment. Median follow-up lasted for 3 months and 35 patients (74.5%), responded to therapy, that is, a decrease in disease activity which lasted for at least a month was noted. Six patients achieved full clinical remission, including five of them a complete endoscopic remission. Furthermore, adjunct in vitro/ex vivo studies investigating effects of naltrexone on intestinal epithelial cells and organoids were performed. Naltrexone significantly reduced endoplasmic reticulum (ER) stress in intestinal tissue organoids, as well as in intestinal epithelial cell cultures exposed to bacteria and bacterial products such as lipopolysaccharide. In a paired test involving epithelial cells obtained from patients before and after LDN treatment, a significantly reduced amount of ER stress was noticed. When subjected to scratch injury, HCT116 and CACO2 colonic epithelial cell cultures treated with naltrexone healed much faster due to increased cellular migration. Though these findings point to a local anti-inflammatory response, systemic levels of cytokines produced by intestinal cells, notably IL-8 and TNF-α, were unchanged in patients on follow-up exams.
Side effects
Low-dose naltrexone for disease prevention and quality of life
Solid evidence for safety and tolerability of chronic LDN is present in the recent Crohn’s trial [12] and MS trial [13], as well as decades of FDA approved daily 50 mg doses for alcoholism. There is no published evidence to support the old ‘‘black box” warning about potential liver damage from chronic high doses [53]. This only happened at extremely high doses that were used in some of the early toxicology trials.
Low Dose Naltrexone: Side Effects and Efficacy in Gastrointestinal Disorders
Side Effect Number of Participants Percentage (%) Neurological Anxiety 19 15.7 Drowsiness 14 11.6 Headache 14 11.6 Dizziness 13 10.7 Insomnia 10 8.3 Muscle pain 10 8.3 Vivid dreams 6 5.0 Mood change 4 3.3 Trouble concentrating 2 1.7 Gastrointestinal Nausea 15 12.4 Abdominal pain 14 11.6 Diarrhea 10 8.3 Anorexia 10 8.3 Other Rash 1 0.1 Hot flashes 1 0.1 Weight gain 1 0.1 Keep in mind that sleep disturbances probably happen because patients were instructed to take it at night. We also have no idea what their lifestyle and diet look like.
Protocol
Low-dose naltrexone for disease prevention and quality of life
Dose Range Dose Specific Mechanism of Action Clinical Use Standard (50–100 mg) Opioid receptor antagonism Alcohol and opiate abuse Low-dose (1–5 mg) Toll-like receptor 4 antagonism, opioid growth factor antagonism Fibromyalgia, multiple sclerosis, Crohn’s disease, cancer, Hailey-Hailey disease, complex-regional pain syndrome Very low-dose (0.001–1 mg) Possibly same as low-dose Add-on to methadone detoxification taper Ultra low-dose (<0.001 mg) Binding to high affinity filamin-A (FLNA) site and reducing μ-opioid receptor associated Gs-coupling Potentiating opioid analgesia There has been considerable confusion into the mechanism of LDN. The most common idea is that it's an opioid antagonist, and the withdrawal of it causes a rebound increase in endorphin, producing the common quality-of-life improvement. This has led people to erroneously believe it will make you feel bad until withdrawal -- so they take it at night. This is a mistake.
I've seen no case reports of people taking LDN during daytime and suffering any negative feelings. If anything, they immediately feel better and more clear. Forgetful people usually do not forget to take their LDN. LDN tends to disrupt sleep if taken too late in the day. Case reports indicate that you can "get over the sleep issues," but what this really means is that you become so deprived of quality sleep that you can fall asleep despite the LDN. It does not have this effect when taken in the morning.
- Start with 1.5mg, and titrate up to 4.5mg (as AgelessRx recommends) if conditions do not improve.
- On a consistent basis, take in in the morning only. - If you are severely injured, take extra doses every ~8 hours until the injury heals. Withdrawal may slow healing, but extra doses accelerate it.
- It may be worth experimenting with 'ULDN,' ultra-low dose naltrexone, if the initial dose is too much.
-
RE: Warm tone light mode for pdfsposted in Bioenergetics Discussion
@CO3 What kind of e ink tablet do you have? Ive been looking into getting one. The onyx book note air 3 looks good, if a bit pricey. Id want something that can run android OS so I can put my Obsidian vault on it.