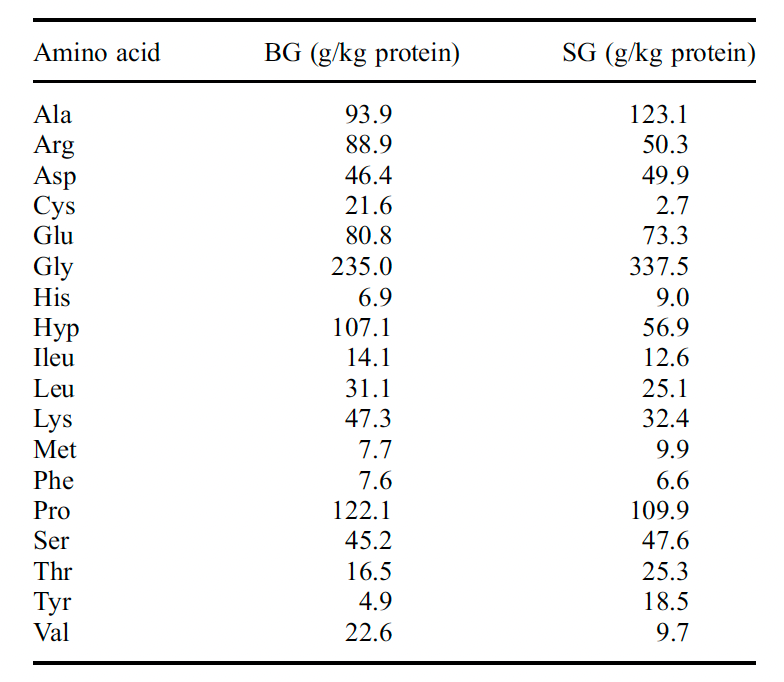
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10321214/
"Inflammatory bowel disease (IBD) is a multifactorial disease with increasing incidence in the U.S. suggesting that environmental factors, including diet, are involved. It has been suggested that excessive consumption of linoleic acid (LA, C18:2 omega-6), which must be obtained from the diet, may promote the development of IBD in humans. To demonstrate a causal link between LA and IBD, we show that a high fat diet (HFD) based on soybean oil (SO), which is comprised of ~55% LA, increases susceptibility to colitis in several models, including IBD-susceptible IL10 knockout mice. This effect was not observed with low-LA HFDs derived from genetically modified soybean oil or olive oil. The conventional SO HFD causes classical IBD symptoms including immune dysfunction, increased intestinal epithelial barrier permeability, and disruption of the balance of isoforms from the IBD susceptibility gene Hepatocyte Nuclear Factor 4α (HNF4α). The SO HFD causes gut dysbiosis, including increased abundance of an endogenous adherent invasive Escherichia coli (AIEC), which can use LA as a carbon source. Metabolomic analysis shows that in the mouse gut, even in the absence of bacteria, the presence of soybean oil increases levels of LA, oxylipins and prostaglandins. Many compounds in the endocannabinoid system, which are protective against IBD, are decreased by SO both in vivo and in vitro. These results indicate that a high LA diet increases susceptibility to colitis via microbial and host-initiated pathways involving alterations in the balance of bioactive metabolites of omega-6 and omega-3 polyunsaturated fatty acids, as well as HNF4α isoforms."
So genetically removing PUFA from soybean oil, prevents IBD. Omega-6 is a carbon source for adherent invasive Escherichia coli. Another study found that this bacteria can cause Parkinson:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10730176/
"Markedly, the relative abundance of E. coli in the LRRK2+/sPD+ group was almost seven times higher than that in the LRRK2+/sPD- group."
So people with a genetic risk variant of Parkinson's (a gene that is involved in the intestinal epithelial barrier), but who don't get Parkinson, have 7 times lower levels of E. coli.
"Animal experiments showed that E. coli administration triggered pathological α-syn accumulation in the colon and spread to the brain via the gut-brain axis in Lrrk2 R1628P mice, due to the co-occurrence of Lrrk2 variant-induced inhibition of α-syn autophagic degradation and increased phosphorylation of α-syn caused by curli in E. coli-derived extracellular vesicles. Fecal microbiota transplantation (FMT) effectively ameliorated motor deficits and α-syn pathology in Lrrk2 R1628P mice. Our findings elaborate on the mechanism that E. coli triggers α-syn pathology in Lrrk2 R1628P mice, and highlight a novel gene-environment interaction pattern in LRRK2 risk variants. Even more importantly, the findings reveal the interplay between the specific risk gene and the matched environmental factors triggers the initiation of α-syn pathology in sPD."
So eating a lot of PUFA from seed oil gives you not only a risk of IBD, but also Parkinson's.
Here is a study of another problem with PUFA. We are constantly producing ROS in the mitochondria, and we know from animals that have a lot of PUFA near there mitochondria, they don't live as long as animals that favor more saturated fats near there mitochrondia. This is a study that Ray Peat mentioned in his last newsletter:
https://sci-hub.se/10.1016/j.bbabio.2006.01.009
"ROS can damage many different kinds of cellular macromolecules including lipids, proteins and DNA. Damage to DNA
may be the most important for aging because it can lead to
irreversible loss or alteration of genetic information in postmitotic cells. mtDNA is situated very close to or even in
contact with the site of mitochondrial ROS production. Since
long-lived vertebrates have low rates of MitROS generation,
this should affect the level of oxidative damage and somatic
mutations in their mtDNA. In agreement with this notion it has
been found that brain and heart 8-oxodG in mtDNA correlates
negatively with maximum longevity in mammals and birds
[39]. This agrees with the lower urinary excretion of 8-oxo-7,8-
dihydroguanine of long-lived animals [70]. In addition, the
levels of 8-oxodG were around 10 fold higher in mtDNA than in
nDNA in the brain and heart of all the 11 studied mammalian
and bird species studied [39,71], a difference similar to that
observed for spontaneous mutations comparing both DNAs.
That suggests that the flux rates of both ROS attack on and
repair of DNA are much higher in the mtDNA of short-lived
than long-lived animals, and are also much higher in the
mtDNA than in the nDNA of all species irrespective of their
longevity [33]. The higher rate of MitROS production of shortlived animals may be an important cause of their much faster
rate of accumulation of mtDNA mutations during aging. A
similar degree of accumulation of mtDNA mutations occurs
after 70–100 years in humans, after 35–50 years in chimpanzees [72] and after only 2–3 years in mice [73].
Unsaturated fatty acids of cellular membranes are the
macromolecules most susceptible to oxidative damage in
cells, and this sensitivity increases as a function of their number
of double bonds. We have found that the cellular membranes of
long-lived mammals and birds have low degrees of fatty acid
unsaturation in mitochondria and tissues including liver, heart,
skeletal muscle, and kidney, and this constitutively protects
their cellular membranes, proteins and DNA against lipid
peroxidation-derived damage [74–77]; reviewed in [78,79].
This is shown in Fig. 1A for heart phospholipid fatty acids in the
heart of 8 animal species with different MLSP [76]. The total
number of double bonds (DBI = double bond index) was
negatively correlated with maximum longevity (Fig. 1A) and
the same was true for the sensitivity to lipid peroxidation (not
shown). Similar results were obtained in liver tissue [77] and
liver mitochondria and their phospholipid classes [75,80] in
different mammalian species, as well as in birds (longer-lived)
compared to mammals of similarly body size. Long-lived
animals obtain a low DBI mainly by avoiding highly
unsaturated fatty acids like 22:6n-3, and sometimes 20:4n-6,
substituting them mainly for 18:3n-3 and 18:2n-6. This lowers
their degree of unsaturation without changing the total amount
of polyunsaturated fatty acids, allowing a strong decrease in
sensitivity to lipid peroxidation probably without major changes
in membrane fluidity, a “homeoviscous-longevity adaptation”
[78]. On the other hand, lipid peroxidation generates aldehydic
products, like malondialdehyde (MDA) and others, that
covalently attach to protein lysine residues. Recently we
found that the level of MDAL protein adducts negatively
correlates with maximum longevity in the heart of different
mammalian species (Fig. 1B) [81]. Thus, the longer the life span
of a species, the lower is its fatty acid double bond content, its
sensitivity to lipid peroxidation and its level of lipoxidationderived protein modification. Bird (long-lived) species also
show lower fatty acid unsaturation and sensitivity to lipid
peroxidation in liver and heart mitochondria (reviewed in [78]),
lower levels of aminoadipic and glutamic semialdehydes
(specific protein carbonyls) and lower levels of CML, CEL
and MDAL in brain proteins than the corresponding comparable
mammals [82]. The relationship between fatty acid unsaturation
and oxidative damage is also observed in experimental studies in
vivo. We have recently observed that treating rats with dietary
oils with a low number of double bonds lowers the degree of
fatty acid unsaturation of brain cellular membranes and lowers
brain lipoxidation-derived protein modification and 8-oxodG
levels in brain mtDNA [83]. This makes sense because lowering
fatty acid unsaturation decreases the susceptibility of membranes to lipid peroxidation. Lipid peroxidation-derived endproducts (“enals”) can also react at the exocyclic amino groups
of dG, dA, and dC to form various alkylated products[84]. Some
common enals that cause DNA damage, analogously to proteins,
are MDA, acrolein, and 4-hydroxynonenal, among others.
Common adducts arising from enals are exocyclic adducts such as etheno adducts, and M1dG. These DNA damage markers are
mutagenic, carcinogenic, and have powerful effects on signal
transduction pathways. Furthermore, they (a) are present in the
genome of healthy humans and other animal species; (b) are
efficient premutagenic lesions that induce mutations frequently
detected in oncogenes or tumor suppressor genes from human
tumors; (c) show increased levels in aged animals; and (d)
!!increase nearly 20 fold with a high PUFA diet!!. Thus, lipid
peroxidation may be a significant endogenous source of DNA
damage and mutations."