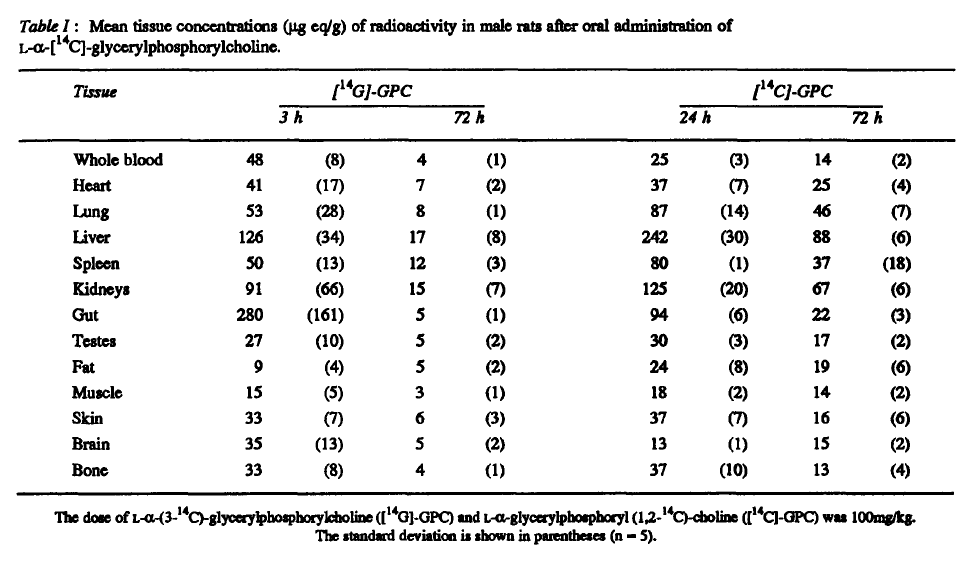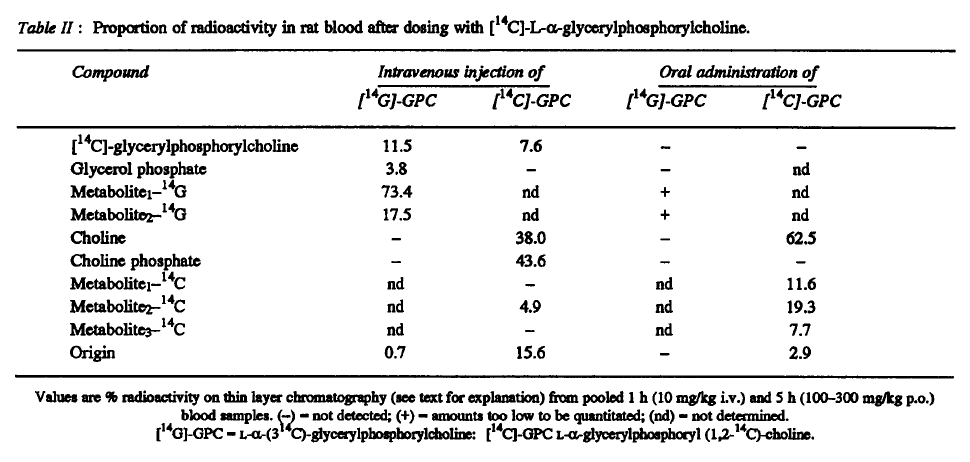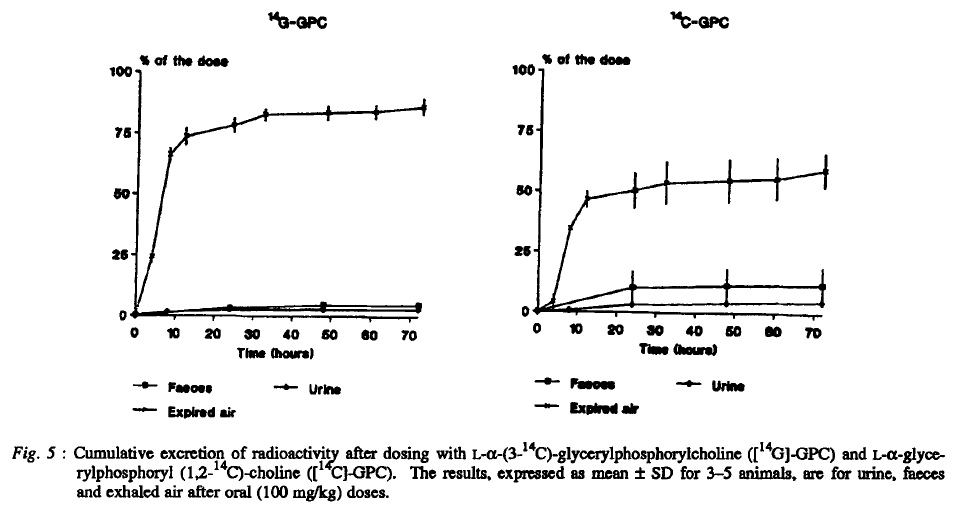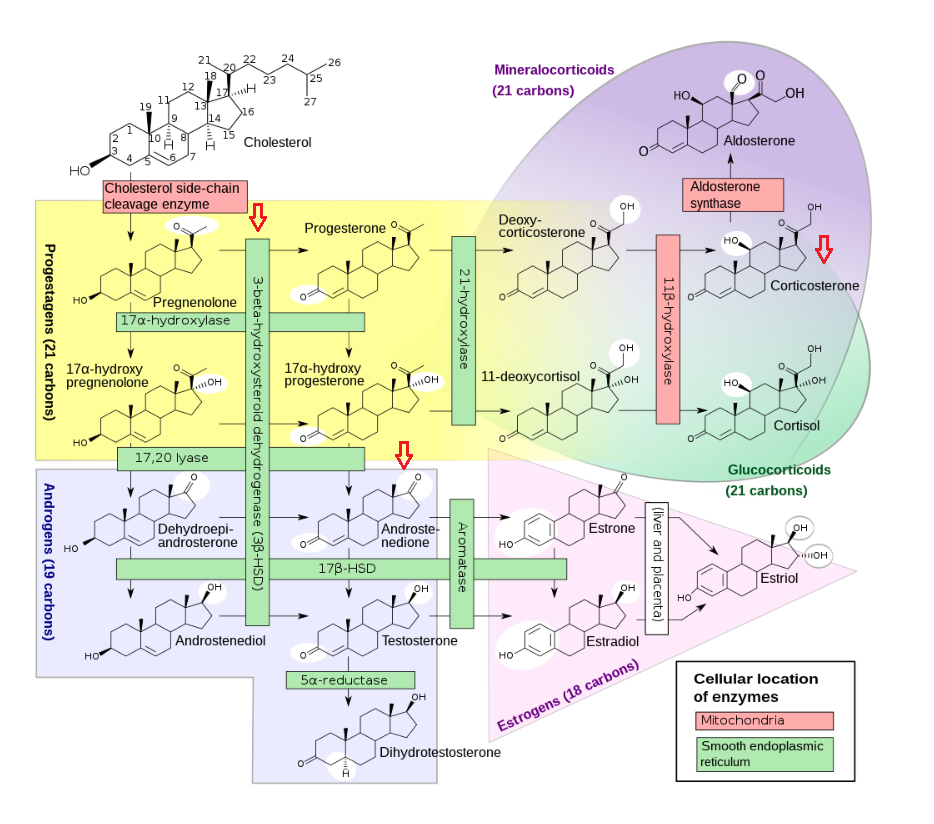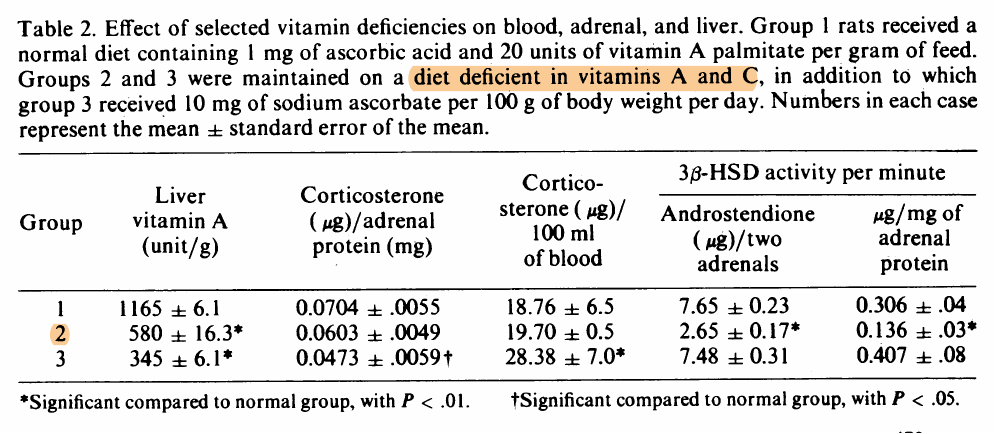
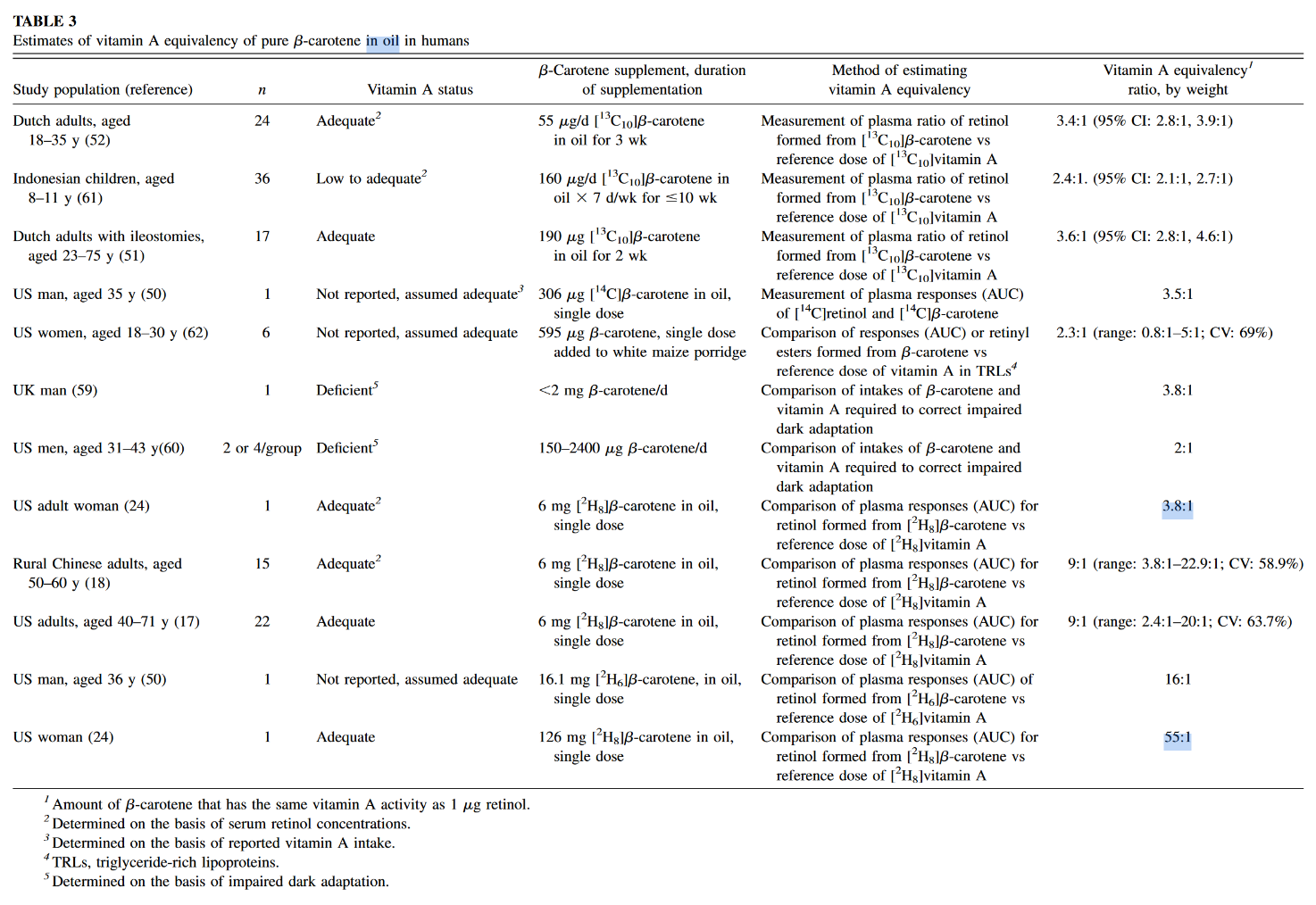
It directs to..
a) Professor Smith:
"If a person takes in more toxic "vit" A than they excrete on a regular basis, then they are ACCUMULATING it in their liver and bodyfat
If 306 mcg beta-carotene (that's 1020 IU, one carrot has 6000 IU!) took 58 DAYS to turn over...do you realize just how fast you are filling up with this toxin?
1 beta-carotene eventually has to split into 2 retinaldehydes"
b) Disciple:
"Wow.
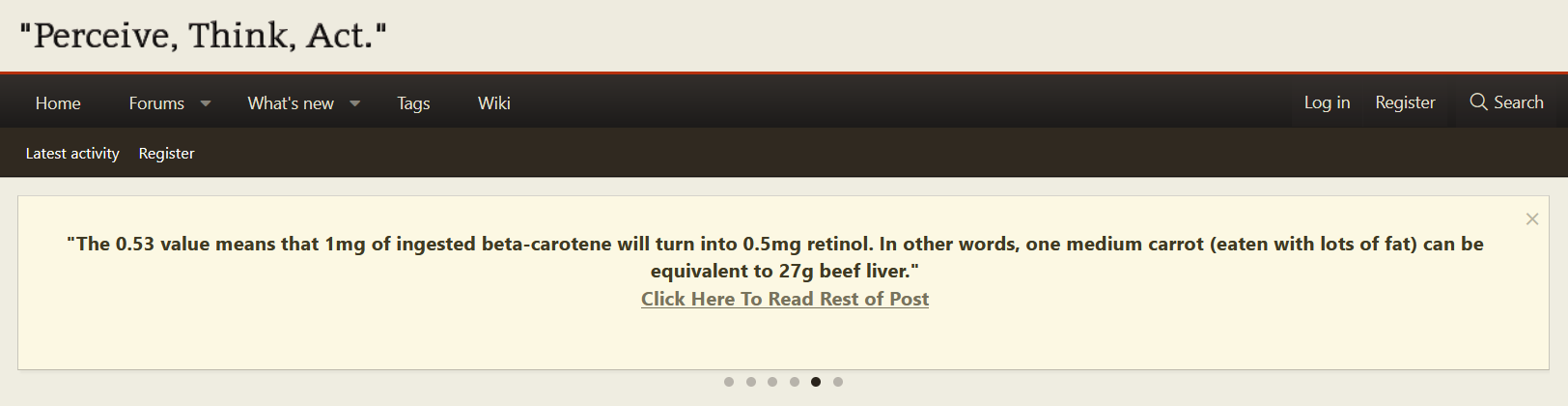
And this is also a great study for showing how much beta-carotene is actually converted to retinol. They write a minimum of 62% is turned into retinol. So a lot will eventually go the retinol route.
"What is clear is that, by our estimates, a minimum of 62% of the absorbed β-carotene was cleaved to vitamin A and by this reasoning the vitamin A value of β-carotene dose was 0.53. [The calculation is as follows: Assuming central cleavage, 1 mol of β-carotene yields 2 mol of vitamin A. If 62% of the absorbed β-carotene were cleaved to vitamin A, 1 mol of β-carotene would yield 1.24 mol of vitamin A. Thus, the vitamin A value of the absorbed dose is 1.24. Yet, the dose was 42.6% absorbed. So, the actual vitamin A value is (1.24) (0.426) or 0.53.]"
1 molecule of beta-carotene turns into 2 molecules of "active vitamin A".
The 0.53 value means that 1mg of ingested beta-carotene will turn into 0.5mg retinol. In other words, one medium carrot (eaten with lots of fat) can be equivalent to 27g beef liver.
Due to the time-delayed conversion, an acute poisoning is not possible, but a chronic poisoning is."
Their claims:
- a) "[..]one carrot has 6000 IU!" (1,800 mcg)
- b) "[..]one medium carrot (eaten with lots of fat) can be equivalent to 27g beef liver."
Beef liver:
- Poisonol 10,000 mcg/100 g
Carrot:
- Alpha-carotene: 2,000 mcg/carrot
- Beta-carotene: 5,000 mcg/carrot
We can disconsider half of the alpha-carotene amount and treat the other half as beta-carotene:
Beef liver:
- Poisonol 10,000 mcg/100 g
Carrot:
- Beta-carotene: 6,000 mcg/carrot
The author of the second post seems to be into the topic of poison A for a while, yet for some reason acts as if the information presented in the mentioned experiment was novel. These equivalences are found in almost all textbooks, even the main sections of the dedicated Wikipedia page have them, suggesting that pure b-macabrotene has half of the activity of poisonol:
- 1 µg RAE = 1 µg retinol from food or supplements
- 1 µg RAE = 2 µg all-trans-β-carotene from supplements
Now the equivalences from their claims:
- a) 1 µg RAE = 3.3 µg of all-trans-β-carotene from carrots
- b) 1 µg RAE = 2 µg of all-trans-β-carotene from carrots
The disciple is basing his equivalence on the following values from the linked experiment:
- Absorption: 42.6%
- Conversion: 62%
- Total (42.6% × 62%): 26%
This is close to a quarter, that results in a mass equivalence of about poison 1:4 b-mac, not 1:2 as he's arguing.
The source of confusion is because he's applying a molar to a mass ratio. B-carotene can yield two poisonols, but the mass is going to remain similar after cleavage and metabolism, with minor gains from the incorporation of new elements (that I'm going to disregard).
To arrive on his value:
- 26% × 2 = 53% (0.53)
It would've been preferable for the disciple to state:
"The 0.53 value means that 1 mg of ingested beta-carotene will turn into
0.5mg~0.25 mg retinol." The factor accounted for the potential of b-carotene to yield two poisonols, so it has to be applied after we halve the amount.
At this stage, we can already discard the idea that a carrot will poison you as an ounce beef liver.
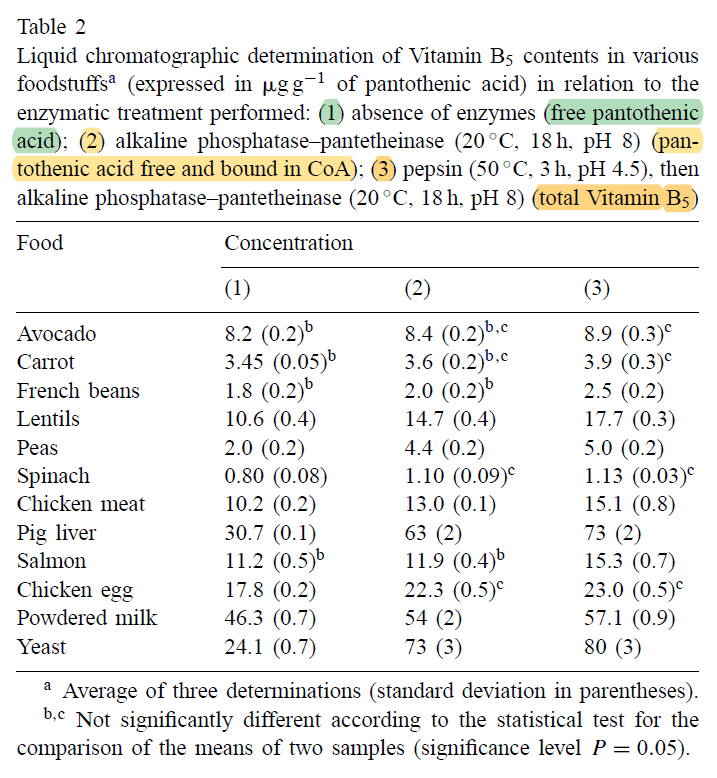
The spinach used in the quoted experiment served to tag macabrotene, the plant assimilated the labeled carbon of CO2 from the environment, allowing them to track the poison. But when it was time to contaminate the victim, the macabrotene was isolated and given in purified form at a low dose.
The food matrix lowers the availability of toxins, making them more difficult to be extracted, which is why researchers define a marked drop in efficiency when the b-macabrotene is derived from foods:
- 1 µg RAE = 1 µg retinol from food or supplements
- 1 µg RAE = 2 µg all-trans-β-carotene from supplements
- 1 µg RAE = 12 µg of all-trans-β-carotene from food
If we ignore this factor and pretend that fibrous carrots are an oil, the amount used in the experiment was modest (300 mcg) and we know that the rate of absorption and conversion tends to decrease as the dose gets high.
But let's reject this too and assume that it remains uninhibited up to 6 mg or so. However, the adjustment is beyond the meal content, the degree of body contamination with the poison is another factor that determines its fate. It's why the purity of the victim matters to interpret the dosing response to carotenoids: contaminated and decontaminated persons will metabolize it differently. When the body is overloaded, the rate of conversion to retinoids is reduced. To a lesser extent, the same goes for their absorption. Yet, carotenoids can be excreted intact.
"1 beta-carotene eventually has to split into 2 retinaldehydes"
It doesn't. His own equivalence conflicts with this claim, even if we discount the unabsorbed fraction (and he probably didn't):
"1 beta-carotene eventually has to split into 2 retinaldehydes":
- 6,000 mcg b-carotene → about 6,000 mcg PAE
"1 [absorbed] beta-carotene eventually has to split into 2 retinaldehydes":
- 6,000 mcg b-carotene → about 2,500 mcg PAE
"[..]one carrot has 6000 IU!":
- 6,000 mcg b-carotene → about 1,800 mcg PAE
Which is it?
"If you take something more than you can excrete, you accumulate"
Of course.
"If 306 mcg beta-carotene (that's 1020 IU, one carrot has 6000 IU!) took 58 DAYS to turn over...do you realize just how fast you are filling up with this toxin?"
A long half-life doesn't result in an indefinite accumulation, only until a steady state is reached, when poisoning for profits should plateau with regular consumption. Something like this in dosing more often:
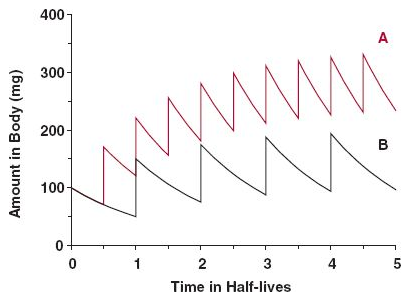
Source: the internet.
Some accumulation is normal and healthy; or should we treat macabrotenoids as toxins to be nuked from the body at all costs? "Vitamin" E is a toxin too (Smith, 2023), but is it harmful when it's not detoxified fast enough to avoid a detectable level, leading to 'accumulation' to be associated with problems?
Consider this: you consume a meal, whose elimination would take 24 hours. Instead of waiting for this period to eat the next, you consume multiple meals in between (may be 4 in a day), that result in accumulation of matter, but without issues. The accumulation is temporary, purposeful and the body is adapted to it. It's not immediate, but the matter eventually leaves the body to the extent that it's consumed: no stuffing until explosion occurs because the input matches the output despite the delay.
Excess carotenoids or calories should come with signs. The early stages are benign, will indicate that the person has to cut back or improve the capacity to handle them. Yet, carotenemia isn't reliable to diagnose poison A toxicity because it can be caused for different reasons (such as lack of nutrients or poor protein synthesis). As an example, if a person needs to take supplemental thyroid hormones and suddenly stops the treatment, it can be followed by carotenemia.
The rate of storage and mobilization is adaptable as well. In case someone discontinues the consumption of poisons altogether, it can signal conservation and a longer persistence in the body. Therefore, the length is variable.
All in all, while we have people debating whether an inability to convert macabrotenes is common or a real concern for persons who don't consume preformed poisons, we have the poison A crowd alarming you that regular carrot consumption is a ticket to the hospital over time, for having the same potential of destruction as liver in the diet, neglecting the multiple steps that regulate these toxins as drugs. #toxicbileapocalypse