On retinaldehyde metabolism
Molecular weights:
- Glucose: 180 g per 1 mol
- Glyceral(dehyde): 90 g per 1 mol
- Retinol: 290 g per 1 mol (290 μg per 1 μmol)
A person may consume 1 mol of glucose (180 g) in a day and complain of issues with retinol while ingesting 1 μmol (290 μg) of it.
Glucose is split at some stage of its oxidation:
- 1 glucose → 2 glyceral(dehyde 3-phosphate)
In the situation above, the body would have to process 2 mol of aldehydes in a day from glucose alone.
Glyceral dehydrogenase and retinal dehydrogenase depend on NAD+ for oxidation.
It's possible that the ratio of NAD+ to NADH affects the metabolism of poisons in disfavor of oxidation, but the body has enough NAD+ to metabolize..
- 2,000,000 μmol of glyceral
Yet not to metabolize..
- 1 μmol of retinal when needed?
Retinal dehydrogenase would have to be way down the priority rank to get its meager share of NAD+ only after other needs are met. It could have evolved strategically this way to function as a 'nutrostat' and become more active to produce (the regulatory) retinoic acids depending on nourishment state.
However, vitamin A is also involved in the induction of genes related to differentiation. It has to be functional in a phase that's marked by anabolism and likely higher levels of NAD(P)H. If its oxidation was heavily dependent on the oxidized:reduced ratio of NAD, it could be a problem.
If retinoic acids are short-lived and the poison A crowd assume to be stuck with retinal because of inability to process it due to NAD+ insufficiency, how would they explain the elevated level in inflammed tissues, found in the conditions that afflict them?
- Retinoic Acid Is Elevated in the Mucosa of Patients With Active Ulcerative Colitis and Displays a Proinflammatory Role by Augmenting IL-17 and IFNγ Production
- Increased Production of Retinoic Acid by Intestinal Macrophages Contributes to Their Inflammatory Phenotype in Patients With Crohn's Disease
Interestingly, it's by impairing the conversion of retinal to retinoic acids that some researchers could get the immunosuppressive effect that the scaremongers seek to recreate with deprivation:
Retinal dehydrogenase can become saturated, but when its capacity is exceeded or when it's inactive, the body can rely on the generic aldehyde oxidase to metabolize retinal and prevent its accumulation, and it doesn't depend on NAD+. Therefore, NAD+ is important, but not essential for the conversion of retinal to retinoic acid.
"Cytosolic enzymes appear to be responsible for the overwhelming majority of atRA formation from at-retinal [28–31]. The ALDH enzymes along with AOX and XOX are cytosolic soluble enzymes that form atRA from at-retinal [32, 33]. While ALDH enzymes are NAD(P)+ dependent, AOX and XOX do not require a pyridine nucleotide cofactor for their activity [17, 33], and the necessity of NAD(P)+ for the majority of atRA formation in the mouse liver was used to show that ALDH enzymes contribute to the majority of atRA formation [30]. An AOX inhibitor, pyridoxal, only inhibited approximately 5% of the atRA formation. Similarly, 80% of the atRA formation in human liver cytosol was NAD+ dependent and the overall contribution of AOX was approximately 10% [31]. Whether microsomal NADPH dependent enzymes such as cytochrome P450s contribute to in vivo atRA synthesis is poorly studied. This is mainly because the Km values for at-retinal with cytochrome P450 are nearly 70-fold higher than endogenous concentrations of at-retinal suggesting they most likely do not have a significant role in atRA biosynthesis [16, 24]. In contrast, the Km values for at-retinal with ALDH1A1-1A3 are low nanomolar [34]. The ALDH1A enzymes also catalyze the formation of atRA from at-retinal bound to cellular retinol binding protein 1 (CRBP1) which is expressed ubiquitously across the body [35–37]."
Aldehyde Oxidase Contributes to All-Trans-Retinoic Acid Biosynthesis in Human Liver
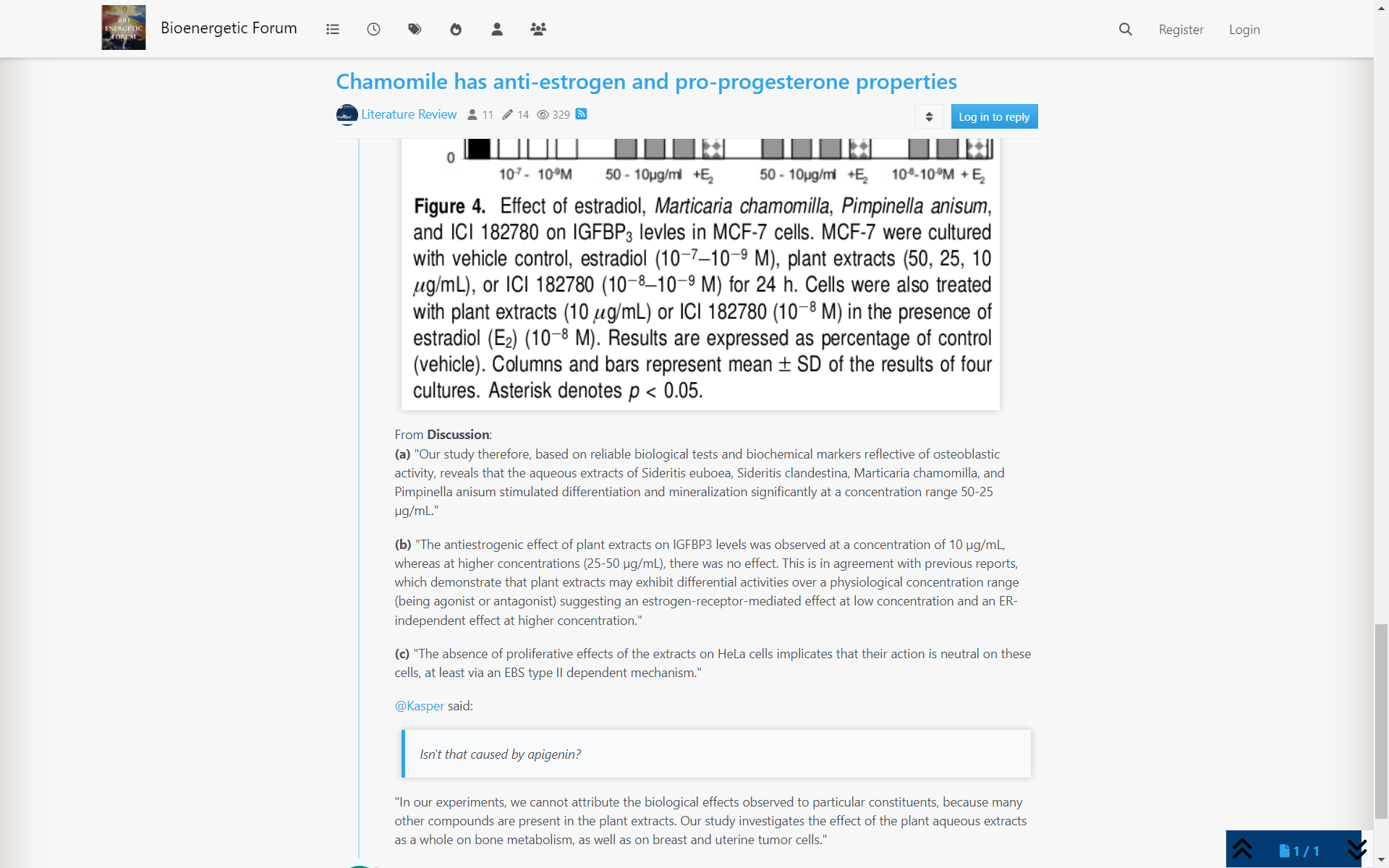
"This study unequivocally shows that AOX forms atRA from retinaldehyde and that in human liver ALDH1A1 and AOX contribute to atRA biosynthesis. Recombinant AOX exhibited about 6-fold higher Km and 1.6-fold higher CLint than ALDH1A1 for atRA formation, showing that ALDH1A1 is a high-affinity, low-capacity atRA synthesizing enzyme, whereas AOX is a low-affinity, high-capacity enzyme. According to the human protein atlas data base, AOX protein is expressed in the kidney, bladder, pancreas, endocrine tissues, and reproductive tissues. This suggests that AOX may contribute to atRA synthesis in the testis, among other tissues. However, studies in healthy mice have shown that AOX activity is not quantitatively significant in the mouse testis (Arnold et al., 2015a) but rather that AOX plays a role in testicular atRA synthesis in mice when ALDH1A activity is impaired (Beedle et al., 2019). This is in agreement with the findings in the human testis showing that ALDH1A1 and ALDH1A2 are responsible for >95% of atRA formation in this tissue (Arnold et al., 2015). In contrast to the testis, AOX appears to contribute about 50% of atRA biosynthesis in mouse liver (Arnold et al., 2015a). This suggests a tissue-specific role of AOX that likely depends on the expression levels of AOX and the complement of ALDH1A enzymes expressed in the different tissues. The results of this study support a similar role of AOX in atRA biosynthesis in human liver as in the mouse liver, with AOX contributing between 33% and 70% of atRA biosynthesis depending on retinaldehyde concentration."
"The reported total concentrations of retinaldehyde in the liver and adipose are 100–200 pmol/g (~150 nM) (Ziouzenkova et al., 2007; Kane et al., 2008). These concentrations are much below the Km toward AOX (1.5 mM) but similar to the Km toward ALDH1A1 (0.25 mM). Because of the difference in Km values for AOX and ALDH1A1, the predicted and observed fm values of retinaldehyde by AOX show a clear dependence on retinaldehyde concentration within the evaluated range of 20–2,000 nM retinaldehyde. As the concentration of retinaldehyde increases, fm,AOX increases due to the saturation of the high-affinity ALDH1A1."
"This also suggests that in the case of ALDH1A dysfunction (inhibition, downregulation, or genetic defect) or excess retinoid/vitamin A (saturation of ALDH1A1), AOX plays an important role in regulating hepatic vitamin A homeostasis. The relative contribution of AOX (30%–70%) to atRA biosynthesis is supported by the scaling of atRA formation from recombinant enzymes, the quantification of AOX and ALDH1A1 protein expression in the liver, the cofactor dependence of atRA formation, and the inhibition of atRA formation by selective ALDH1A and AOX inhibitors WIN18,446 and hydralazine. These tools developed in the current study allow differentiation of ALDH1A and AOX contributions to atRA formation and can be further applied to other critical retinoid responsive tissues, such as the skin, hematopoietic cells, and adipose tissue, to determine the enzymes contributing to atRA biosynthesis. Such information will be useful in predicting how inhibition or genetic variability in AOX and ALDH1A enzymes will affect retinoid homeostasis and signaling."
Two different enzymes are primarily responsible for retinoic acid synthesis in rabbit liver cytosol
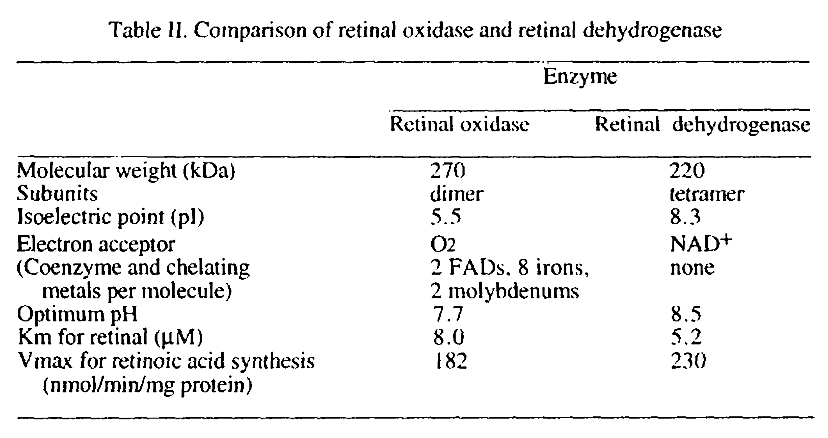
Values aren't reliable.
The preferred route doesn't depend on molybdenum. The alternative route result in formation of hydrogen peroxide, but the amounts shouldn't be monstrous, although it could occur at susceptible sites.
Choline isn't used up in esterifying retinol
If retinal couldn't be oxidized by any means, it could be converted to retinol for temporary storage.
The enzymes involved in esterification are acyltransferases:
- Lecithin:retinol acyltransferase (also high-affinity, low-capacity)
- Acyl-CoA:retinol acyltransferase
Genetic Variations Associated with Vitamin A Status and Vitamin A Bioavailability
"LRAT uses fatty acids from intracellular membrane phospholipids, while DGAT1 and the other ARAT(s) use newly-absorbed fatty acids[.]"
Phosphatidylcholine functions as a fatty acid donor and retinol would be the acceptor (to yield retinyl palmitate, stearate, oleate..). What's being consumed is the fatty acid of a structural lipid, not its choline. After the reaction, the lost fatty acid of phosphatidylcholine can be replenished and the molecule will be renewed.
If a lack of choline is severe enough to compromise cell structure, the person has greater concerns than the esterification of retinol. And the body can switch to the alternative pathway.
Free retinol appears in circulation after meals with higher vitamin A doses, showing that the esterification process can be overwhelmed, but it can be prevented by avoiding equine amounts at once and by consuming extra fat with the meal to cooperate with the backup acyltransferase.